Endokrynol. Ped. 12/2013;3(44):9-16
DOI: 10.18544/EP-01.12.03.1454
Diagnostyka i leczenie heterozygotycznej postaci hipercholesterolemii rodzinnej u dzieci – doniesienie wstępne
1Katedra i Klinika Pediatrii, Endokrynologii i Diabetologii, Gdański Uniwersytet Medyczny
2I Katedra i Klinika Kardiologii, Gdański Uniwersytet Medyczny
Słowa kluczowe: hipercholesterolemia, dzieci, LDLR, APOB
Streszczenie
Hipercholesterolemia rodzinna (familial hipercholesterolemia, FH) jest najczęstszą chorobą monogenową. Przyczyną FH są dziedziczone autosomalnie dominująco mutacje w genie kodującym receptor LDL, apolipoproteinę B (APOB) lub PCSK9. Celem pracy jest analiza danych klinicznych dzieci i młodzieży z potwierdzoną badaniem molekularnym heterozygotyczną postacią hipercholesterolemii rodzinnej, będących pod opieką Katedry Pediatrii, Diabetologii i Endokrynologii GUM, oraz wstępna ocena efektów leczenia. Pacjenci i metody. Do badania włączono dzieci z podwyższonym poziomem cholesterolu. U wszystkich pacjentów wykluczono wtórne przyczyny hipercholesterolemii. U 127 pacjentów zlecono badanie molekularne w kierunku mutacji w genie receptora LDL oraz Apo B. Wyniki. Obecnie pod opieką Kliniki Pediatrii, Diabetologii i Endokrynologii znajduje się 43 pacjentów z potwierdzoną genetycznie FH oraz ponad 27 pacjentów z podejrzeniem hipercholesterolemii rodzinnej w trakcie badań molekularnych. U wszystkich pacjentów z FH wywiad rodzinny w kierunku chorób układu sercowo-naczyniowego był dodatni. W momencie zgłoszenia się do Kliniki średni poziom cholesterolu całkowitego pacjentów wynosił 281,6±61,6mg/dl, poziom cholesterolu LDL 205,24±70,51 mg/dl. Badania genetyczne wykazały u 74% pacjentów mutacje w genie receptora LDL, a u 26 % mutacje w genie Apo B. Leczenie preparatem statyn włączono u 27 pacjentów oraz ezetimibem u 2 pacjentów z FH. Średni poziom cholesterolu całkowitego w badaniu kontrolnym po 6–8 tygodniach leczenia wynosił 203,7±38,1 mg/dl, cholesterolu LDL 128,9±43,4 mg/dl. W grupie pacjentów leczonych farmakologicznie nie obserwowano działań niepożądanych stosowanej terapii. Zauważono, że u osób leczonych farmakologicznie grubość kompleksu intima-media (IMT) w tętnicach szyjnych była niższa w porównaniu z dziećmi leczonymi wyłącznie dietą. Wnioski. Zaburzenia lipidowe powinny być rozpoznawane i leczone od jak najwcześniejszych lat życia, szczególnie u dzieci z obciążonym wywiadem rodzinnym w kierunku chorób układu sercowo-naczyniowego. Obecnie najbardziej skutecznym leczeniem wśród dzieci wydaje się terapia statynami. Konieczna jest dalsza obserwacja i monitorowanie skuteczności i bezpieczeństwa terapii statynami
Wstęp
Hipercholesterolemia rodzinna (familial hipercholesterolemia, FH) jest najczęstszą wrodzoną chorobą o podłożu monogenowym [1, 2]. Wyróżnić można dwie postaci hipercholesterolemii rodzinnej: heterozygotyczną występującą z częstością 1:500 osób oraz homozygotyczną występującą z częstością 1:1 000 000. Hipercholesterolemia rodzinna jest dziedziczona w sposób autosomalny dominujący i najczęściej spowodowana jest mutacją w genie kodującym receptor LDL (LDLR), apolipoproteinę B (APOB) lub PCSK9 [3–6].
Charakterystyczne dla hipercholesterolemii rodzinnej podwyższone stężenie cholesterolu stanowi przyczynę wczesnego rozwoju miażdżycy naczyń oraz wynikającego z niej zwiększonego ryzyka chorób układu sercowo-naczyniowego już w młodym wieku. Wczesne wykrycie hipercholesterolemii rodzinnej, jeszcze w okresie dzieciństwa, pozwala na szybszą modyfikację stylu życia oraz wdrożenie leczenia farmakologicznego, które pozwoli w przyszłości zmniejszyć ryzyko wystąpienia miażdżycy oraz jej powikłań. W ciągu ostatnich lat dzięki możliwości wykonywania badań molekularnych i poszukiwania mutacji odpowiedzialnych za FH coraz częściej hipercholesterolemia rodzinna jest rozpoznawana w młodym wieku, co daje możliwość wczesnego, skutecznego leczenia i pozwala zapobiegać rozwojowi chorób układu sercowo-naczyniowego. Frakcja cholesterolu o niskiej gęstości (Low Density Lipoprotein, LDL) pochodzi z pożywienia oraz z endogennej syntezy zachodzącej głównie w hepatocytach. Cholesterol frakcji LDL łączy się z receptorami LDL znajdującymi się na hepatocytach i w ten sposób jest usuwany z krążenia [1]. Najczęstszą przyczyną FH jest mutacja w genie receptora LDL, dotychczas w literaturze opisano ponad 1700 mutacji w tym genie [7]. Z uwagi na jego ekspresję można podzielić hipercholesterolemię rodzinną na pięć klas, gdzie klasa I oznacza całkowity brak ekspresji receptora, a klasa V brak zdolności receptora do transportu LDL. Pozostałe klasy są stanem pośrednim między klasą I a klasą V [8]. W wyniku upośledzonego usuwania cząsteczek LDL z osocza wzrasta ich stężenie w surowicy krwi, co sprzyja odkładaniu się cholesterolu w ścianie naczyń i prowadzi do rozwoju miażdżycy.
Nagromadzenie cholesterolu w tkankach może dawać objawy charakterystyczne dla chorych z hipercholesterolemią rodzinną, takie jak żółtaki płaskie powiek, żółtaki ścięgien Achillesa, żółtaki guzowate, rąbek starczy rogówki, objawy miażdżycy naczyń, jednak oznaki takie praktycznie nie występują u dzieci i młodzieży z postacią heterozygotyczną hipercholesterolemii rodzinnej [9].
U pacjentów z dodatnim wywiadem rodzinnym w kierunku hipercholesterolemii lub przedwczesnych zdarzeń sercowo-naczyniowych badania przesiewowe w kierunku FH należy rozpoczynać już po drugim roku życia, kiedy następuje stabilizacja parametrów gospodarki lipidowej [10–12]. W trakcie przeprowadzania badań diagnostycznych należy zawsze wykluczyć wtórne przyczyny podwyższonego poziomu cholesterolu, takie jak: niedoczynność tarczycy, choroby nerek, celiakię lub choroby wątroby, a także przyjmowanie niektórych leków, tj. diuretyków tiazydowych i pętlowych, kortykosteroidów, cyklosporyny, estrogenów, progesteronu, inhibitorów proteazy [13, 14].
Hipercholesterolemię rodzinną należy podejrzewać u dzieci i młodzieży (<20 roku życia), u których poziom cholesterolu frakcji LDL wynosi powyżej 160 mg/dl [2]. Badanie profilu lipidowego należy powtórzyć po włączeniu diety z niską zawartością cholesterolu. Kryteria rozpoznania hipercholesterolemii rodzinnej zostały zaproponowane przez trzy niezależne grupy badawcze: The Simon Broom Register Group z Wielkiej Brytanii, MedPed Program ze Stanów Zjednoczonych oraz The Duch Lipid Clinic Network z Holandii. We wszystkich zaproponowanych kryteriach pod uwagę brane jest stężenie cholesterolu całkowitego, natomiast kryteria utworzone przez grupę Simona Brooma oraz przez ekspertów WHO (The Duch Lipid Clinic Network) podkreślają także istotne znaczenie wywiadu, historii rodzinnej, a także badania genetycznego w postawieniu ostatecznego rozpoznania FH [7, 15].
Cel pracy
Celem pracy była analiza przypadków dzieci i młodzieży z potwierdzoną badaniem molekularnym hipercholesterolemią rodzinną, będących pod opieką Kliniki Pediatrii, Diabetologii i Endokrynologii Gdańskiego Uniwersytetu Medycznego oraz Krajowego Centrum Hipercholesterolemii Rodzinnej, a także ocena uzyskanych wyników badań oraz wstępna ocena efektów wprowadzonego leczenia.
Pacjenci i metody Do badania włączono pacjentów z hipercholesterolemią będących pod opieką Poradni Zaburzeń Lipidowych przy Klinice Pediatrii, Diabetologii i Endokrynologii Gdańskiego Uniwersytetu Medycznego. U wszystkich pacjentów zostały wykonane badania biochemiczne: stężenie cholesterolu całkowitego i jego frakcji LDL i HDL oraz trójglicerydów, apolipoproteiny A i B, kreatyniny, enzymów wątrobowych, hormonów tarczycy, glukozy, hemoglobiny glikowanej oraz przeciwciał przeciw transglutaminazie tkankowej. Grupę 127 pacjentów z hipercholesterolemią, po wykluczeniu jej wtórnych przyczyn, poddano badaniu molekularnemu celem poszukiwania mutacji w genie receptora LDL (LDLR) oraz genie apolipoproteiny B 100 (APOB). Badanie przeprowadzono w Katedrze i Zakładzie Biologii i Genetyki Gdańskiego Uniwersytetu Medycznego przy użyciu techniki sekwencjonowania bezpośredniego genu LDL receptora i fragmentu egzonu 26 genu apolipoproteiny B100 (APOB) oraz z wykorzystaniem techniki MLPA (Multiplex Ligation-dependent Probe Amplification).
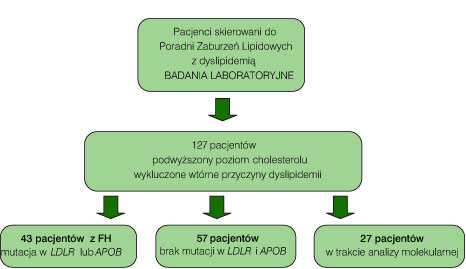
Wyniki
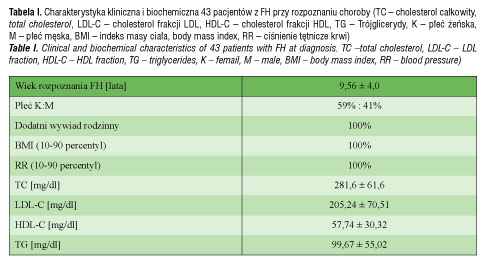
U wszystkich pacjentów z podwyższonym poziomem cholesterolu badania diagnostyczne rozpoczęto od wykluczenia wtórnych przyczyn hipercholesterolemii rodzinnej – niedoczynności tarczycy, zaburzeń w gospodarce węglowodanowej, chorób nerek i wątroby oraz celiakii. Dodatni wywiad rodzinny w kierunku hipercholesterolemii lub chorób układu sercowo-naczyniowego stwierdzono u 100% pacjentów. Nastpnie wykonano badanie molekularne u 127 pacjentów. U 43 badanych dzieci stwierdzono obecność mutacji w genie LDLR lub APOB, u 27 pacjentów wyniki nadal pozostają w opracowaniu, a u 57 dzieci, u których na podstawie objawów klinicznych, wywiadu rodzinnego i wyników badań laboratoryjnych wysunięto podejrzenie hipercholesterolemii rodzinnej, nie stwierdzono mutacji w badanych genach. W grupie pacjentów z dodatnim wynikiem badania genetycznego w 74% przypadków stwierdzono mutację w genie LDLR, a u 26% w genie APOB. W momencie zgłoszenia się do Poradni Zaburzeń Lipidowych średni wiek pacjentów z hipercholesterolemią rodzinną wynosił 9,56 ± 4,0 lata i 41% stanowiły dzieci płci męskiej, a 59% płci żeńskiej. U większości dzieci i młodzieży, skierowanych do Poradni Zaburzeń Lipidowych, badanie poziomu cholesterolu było wykonywane z powodu dodatniego wywiadu rodzinnego w kierunku chorób układu sercowo-naczyniowego lub hipercholesterolemii w rodzinie.
U kilku pacjentów podwyższony poziom cholesterolu został stwierdzony przypadkowo w trakcie kontrolnych badań laboratoryjnych z powodu innych przyczyn lub w trakcie programów zdrowotnych prowadzonych wśród dzieci i młodzieży na terenie województwa pomorskiego. Średni poziom cholesterolu całkowitego u pacjentów pediatrycznych z hipercholesterolemią rodzinną w momencie zgłoszenia się do Poradni Zaburzeń Lipidowych wynosił 281,6 ± 61,6 mg/dl, poziom cholesterolu frakcji LDL 205,24 ± 70,51 mg/dl, poziom frakcji HDL cholesterolu 57,74 ± 30,32 mg/dl oraz poziom trójglicerydów 99,67 ± 55,02 mg/dl. U wszystkich pacjentów z rozpoznaną hipercholesterolemią rodzinną indeks masy ciała (BMI) oraz wartości ciśnienia tętniczego mieściły się w granicach normy dla wieku i płci. U większości pacjentów z genetycznym potwierdzeniem hipercholesterolemii rodzinnej wykonano badanie ultrasonograficzne oceniające grubość kompleksu intima-media (ang. intima-media tickness, IMT) w tętnicach szyjnych. U wszystkich pacjentów z potwierdzoną molekularnie hipercholesterolemią rodzinną uzyskano wynik w górnej granicy normy dla wieku i płci bądź nieco powyżej górnej granicy normy. Średnia wartość kompleksu intima-media tętnicy szyjnej prawej wynosi 0,453 ± 0,04 mm, a tętnicy szyjnej lewej 0,452 ±0,05 mm. Wśród pacjentów 11 miało wykonywane badanie IMT wyłącznie w trakcie leczenia dietą (grubość warstwy środkowej i wewnętrznej tętnicy szyjnej średnio wynosiła 0,461364 ± 0,050156 mm), natomiast 13 pacjentów miało wykonane badanie już w trakcie kilkunastotygodniowego leczenia preparatem statyny (grubość warstwy środkowej i wewnętrznej tętnicy szyjnej średnio wynosiła 0,445769 ± 0,040494 mm). Zauważono, że u osób leczonych farmakologicznie wartość IMT była niższa w porównaniu z dziećmi leczonymi wyłącznie dietą, jednak nie uzyskano istotności statystycznej (ryc. 1).

Wszyscy pacjenci z hipercholesterolemią odbyli szczegółowe szkolenie dietetyczne i na stałe pozostają pod opieką dietetyka. Leczenie preparatami statyn w dawce 5 lub 10 mg włączono u 27 pacjentów oraz z powodu przeciwwskazań do włączenia statyny u 2 pacjentów zastosowano leczenie ezetimibem w dawce 10 mg. Średni wiek pacjentów z hipercholesterolemią rodzinną, którym włączono leczenie preparatem statyny, wynosił 12,26 ± 4,17 lat. Dzieci w wieku poniżej 10 roku życia z rozpoznaną FH były w większości leczone wyłącznie dietą i aktywnością fizyczną oraz substytucją kwasów omega 3. Poziom cholesterolu całkowitego w badaniu kontrolnym po 4–6 tygodniach od włączenia leczenia statyną lub ezetimibem obniżył się średnio o 27 % i wyniósł 203,7 ± 38,1 mg/dl, a cholesterol frakcji LDL o 29% i wyniósł 128,9 ± 43,4 mg/dl.
U wszystkich pacjentów monitorowano także poziom enzymów wątrobowych i kinazy kreatyninowej celem monitorowania leczenia. W ponadrocznej obserwacji leczonych statynami pacjentów poniżej 18 roku życia nie stwierdzono istotnych działań niepożądanych.
Omówienie
Choroby układu sercowo-naczyniowego są jedną z najczęstszych przyczyn zgonów na świecie. Rozwijają się one na podłożu miażdżycy, która przez wiele lat może być bezobjawowa. W grupie przedstawionych pacjentów nie stwierdzono żadnych objawów podwyższonego poziomu cholesterolu w badaniu fizykalnym, co przemawia za bezobjawowym przebiegiem heterozygotycznej postaci choroby w dzieciństwie. Za podstawowy czynnik ryzyka chorób układu sercowo- naczyniowego uznaje się podwyższony poziom cholesterolu LDL. Jedną z przyczyn podwyższonego poziomu cholesterolu może być rodzinna hipercholesterolemia, jednak należy pamiętać o wcześniejszym wykluczeniu wtórnych przyczyn dyslipidemii, m.in. cukrzycy, przewlekłych chorób nerek i wątroby, celiakii oraz niedoczynności tarczycy [1]. W badanej grupie pacjentów, wyselekcjonowanej do wykonania badania molekularnego, w pierwszym etapie wykluczono wtórne przyczyny zaburzeń lipidowych, a grupę pacjentów z potwierdzoną badaniem molekularnym FH charakteryzowały prawidłowe wartości BMI i ciśnienia tętniczego. Jedną z najczęstszych pierwotnych, uwarunkowanych genetycznie dyslipidemii jest hipercholesterolemia rodzinna, która może być wynikiem mutacji w genie LDLR, APOB lub PCSK9. W badanej grupie dzieci, u których stwierdzono molekularne podłoże FH, w 74% przypadków zgodnie z doniesieniami piśmiennictwa były to mutacje w genie LDLR [2]. W grupie dzieci, u których występowała hipercholesterolemia pierwotna, a nie stwierdzono mutacji w badanych genach, zaplanowano poszerzenie diagnostyki w genie PCSK9 w najbliższej przyszłości. Pacjenci ci pozostają pod opieką Poradni Zaburzeń Lipidowych. Z uwagi na ryzyko powikłań sercowo-naczyniowych bardzo ważne jest szybkie wdrożenie terapii hipolipemizującej u osób z FH. U dzieci i młodzieży, u których występuje dziedzicznie uwarunkowana hipercholesterolemia, leczenie dietetyczne jest niewystarczające i w tej grupie pacjentów powinno się rozważyć leczenie farmakologiczne.
Do leków obniżających stężenie lipidów w surowicy krwi należą: żywice wiążące kwasy żółciowe, statyny, niacyna, fibraty oraz inhibitory wchłania cholesterolu. Statyny stały się lekiem pierwszego rzutu preferowanym u dzieci i młodzieży z zaburzeniami lipidowymi [1]. FDA (Food and Drug Administration) dopuściło do ich stosowania u dzieci w hipercholesterolemii rodzinnej. Statyny są jedną z lepiej przebadanych grup leków hipolipemizujących u dorosłych. Jednakże badania u pacjentów pediatrycznych są krótkoterminowe i na tej podstawie nie można przewidzieć odległych konsekwencji ich stosowania [16, 17]. W obserwowanej grupie dzieci i młodzieży nie stwierdzono działań niepożądanych w leczeniu statynami. Początkowa dawka wynosiła 5 lub 10 mg i była skuteczna u większości pacjentów. Dotychczasowe doniesienia wskazują, iż leki te obniżają stężenie cholesterolu LDL nawet o 30–50%, co potwierdzają uzyskane wyniki z prawie 30% redukcją stężenia LDL już po 4–6 tygodniach leczenia w badanej grupie dzieci [16]. Obecna wiedza wskazuje także, iż zastosowana najniższa dawka leku powinna być skuteczną dawką, w przeciwieństwie do pacjentów dorosłych, u których stosuje się leczenie agresywne wysokimi dawkami statyn. Wynika to z różnicy rozwoju miażdżycy u dzieci i dorosłych. U dzieci najczęściej mamy do czynienia z odwracalnym stadium rozwoju miażdżycy, zapobieganiem rozwojowi i dojrzewaniu blaszki miażdżycowej [8, 13], co mogą potwierdzać uzyskane wyniki pomiarów IMT u pacjentów przed leczeniem i w trakcie kilkunastotygodniowego leczenia preparatem statyny. FDA zaleca, aby terapię statyną wdrożyć powyżej 10 roku życia – u dziewcząt, u których wystąpiła już pierwsza miesiączka, oraz u dorastających chłopców (dojrzewanie ocenione w skali Tannera >2). Terapia ta ma być uzupełnieniem leczenia dietetycznego i modyfikacji stylu życia. Leczenie to proponuje się pacjentom, u których stężenie cholesterolu frakcji LDL wynosi powyżej 190 mg/dl albo utrzymuje się powyżej 160 mg/dl mimo prawidłowego postępowania niefarmakologicznego, z wywiadem rodzinnym przedwczesnego występowania choroby wieńcowej, lub stwierdza się u pacjenta dwa lub więcej czynników chorób układu sercowo-naczyniowego [18]. National Lipid Association Expert Panel w zaproponowanym algorytmie postępowania u dzieci i młodzieży wskazuje na konieczność oceny ryzyka sercowo-naczyniowego oraz zastosowanie profilaktyki pierwotnej. Włączenie statyn odbywa się dopiero po modyfikacji stylu życia i nawyków żywieniowych. Leczenie farmakologiczne powinno być włączone po ukończeniu 8 roku życia u pacjentów z heterozygotyczną postacią hipercholesterolemii rodzinnej [19]. Celem terapii jest obniżenie cholesterolu o powyżej 50% oraz redukcja poziomu cholesterolu LDL poniżej 130 mg/dl. Konieczne jest stałe monitorowanie pacjentów celem dobrania odpowiedniej dawki terapeutycznej leku oraz kontroli ewentualnych działań niepożądanych leczenia. Ważnym elementem w terapii chorych z hipercholesterolemią rodzinną jest regularne badanie parametrów biochemicznych. Powinno się kontrolować lipidogram, aby stwierdzić skuteczność terapii farmakologicznej oraz ewentualnie zmodyfikować dawkę leku. U dzieci przyjmujących statyny powinno się oznaczać co 3–4 miesiące enzymy wątrobowe oraz kinazę kreatyniny celem oceny ryzyka wystąpienia działań niepożądanych – toksycznego uszkodzenia wątroby i mięśni. Każdy pacjent powinien być poinformowany o ewentualnych działaniach niepożądanych leku, aby zareagować możliwie szybko w chwili wystąpienia pierwszych objawów. Innym lekiem dopuszczonym do leczenia hipercholesterolemii rodzinnej u dzieci jest inhibitor wchłaniania cholesterolu (ezetimib), który blokuje wchłanianie cholesterolu na poziomie rąbka szczoteczkowego w jelicie cienkim. Ma zastosowanie w przypadku, gdy monoterapia statynami nie przynosi zadowalających rezultatów [8, 19]. U pacjentów pediatrycznych istotne jest także ocenianie wzrostu, przyrostu masy ciała oraz postępu dojrzewania płciowego w trakcie farmakoterapii [9, 19]. Ważnym narzędziem diagnostycznym jest pomiar kompleksu intima-media naczyń tętniczych szyjnych, który w nieinwazyjny sposób pozwala ocenić zaawansowanie zmian miażdżycowych w naczyniach, a następnie monitorować ich ustępowanie pod wpływem leczenia, co jest już zauważalne po 6 miesiącach od wdrożenia terapii statynami [20–22]. W badanej grupie dzieci wykonano badanie IMT i stwierdzono, że pacjenci po włączeniu leczenia charakteryzowali się niższą wartością IMT. Badania IMT są nieinwazyjne i powtarzalne i będą wykonywane raz w roku w grupie pacjentów z rozpoznaną FH celem dalszego monitorowania stanu ściany naczyń i efektów stosowanego leczenia.
Podsumowanie
Zaburzenia lipidowe powinny być rozpoznawane i leczone jak najwcześniej. Wdrożenie odpowiednich badań kaskadowych w rodzinach pacjentów obciążonych hipercholesterolemią rodzinną pozwala na wcześniejsze postawienie diagnozy i podjęcie terapii, która w przyszłości znniejszy ryzyko występowania chorób układu sercowo-naczyniowego w młodym wieku. Oprócz diety oraz modyfikacji stylu życia dzieci i młodzieży z FH należy rozważyć leczenie farmakologiczne. Obecnie najbardziej skutecznym i bezpiecznym leczeniem wydaje się terapia statynami. Niestety z uwagi na krótkotrwałe badania w populacji pediatrycznej wciąż trudno jest przewidzieć ewentualne odległe działania niepożądane tych leków. Dlatego bardzo ważnym elementem w opiece specjalistycznej pacjentów z hipercholesterolemią rodzinną jest częste monitorowanie parametrów biochemicznych oraz wykonywanie pomiarów IMT. Postępowanie takie ma na celu zwolnienie postępu rozwoju miażdżycy, a tym samym zmniejszenie ryzyka chorób układu sercowo-naczyniowego. Konieczna jest dalsza obserwacja i monitorowanie skuteczności i bezpieczeństwa stosowanej terapii u dzieci i młodzieży.
Piśmiennictwo
1. Wierzbicki A.S., Humphries S.E., Minhas R. et al.; Familial hypercholesterolemia: summary of NICE guidance; BMJ 2008:337, a1095
2. Civeira F.; International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia; Atherosclerosis 2004 Mar:173(1), 55-68
3. ; Familial Hypercholesterolemia. A Report of a WHO consultation; WHO Geneva, Switzerland, 1998
4. Innerarity T.L., Weisgraber K.H., Arnold K.S. et al.; Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding; Proc. Natl. Acad. Sci. USA 1987:84, 6919-6923
5. Abifadel M., Varret M., Rab J.P. et al.; Mutations in PCSK9 cause autosomal dominant hypercholesterolemia; Nat. Genet. 2003:34, 154-156
6. Abifadel M., Rabes J.P., Devillers M. et al.; Mutations and Polymorphisms in the Proprotein Convertase Subtilin Kexin 9 (PCSK9) Gene in Cholesterol Metabolism and Disease; Human Mutation 2009:30, 520-529
7. Austin M.A., Hutter C.M., Zimmern R.L. et al.; Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: A HuGE Prevalence Review; Am. J. Epidemiol. 2008:160, 407-420
8. Kwiterovich P.O.; Recognition and management of dyslipidemia in children and adolescents; J. Clin. Endocrinol. Metab. 2008:93 (11), 4200-4209
9. Kubalska J.; Hipercholesterolemia genetycznie uwarunkowana u dzieci, Pediatria Współczesna; Gastroenterologia, Hepatologia i Żywienie dziecka 1999:1 2/3, 129-132
10. Goldberg A.C., Hopkins P.N., Toth P.P. et al.; Familial Hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients. Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia; Journal of Clinical Lipidology 2011:5, 133-140
11. Daniels S.R., Greer F.R. and the Committee on Nutrition; Lipid screening and cardiovascular health in childhood; Pediatrics 2008:122, 198-208
12. National Cholesterol Education Program; Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents; Pediatrics 1992:89, 525-584
13. Reiner Ž., Catapano A.L., De Backer G. et al.; ESC/EAS guidelines for management of dyslipidemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS); Eur. Heart J. 2011:32, 1769-1818
14. Ito M.K., McGowan M.P., Moriaty P.M.; Management of familial hyperlipidemias in adult patients. Recommendations from National Lipid Association Expert Panel of Familial Hypercholesterolemia; J. Clin. Lipidol. 2011:5, 38-45
15. Chmara M.; Mutacje genów LDLR i APOB w hipercholesterolemii rodzinnej; rozprawka doktorska Gdańsk 2005
16. Lamaida N., Capuano E, Pinto L. el al.; The safety of statins in children; Acta Paediatr. 2013 Apr 30. doi: 10.1111/apa.12280
17. Vuorio A., Kuoppala J., Kovanen P.T. et al.; Statins for children with familial hypercholesterolemia; Cochrane Database Syst Rev. 2010 Jul 7, (7)
18. Belay B., Belamarich P., Revzon C.; Stosowanie statyn w pediatrii: podstawy wiedzy, ograniczenia i dalsze kierunki; Pediatria po Dyplomie Grudzień 2008:12, nr 6, 19-31
19. Daniels S.R. et al.; Pediatric aspects of Familial Hypercholesterolemias: Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia; Journal of Clinical Lipidology 2011:5, S30-S37
20. Adamczak-Ratajczak A., Mądry E., Krawczyk M. et al.; Kompleks intima-media – znaczenie diagnostyczne; Family Medicine & Primary Care Review 2010:12, 3, 877-878
21. Wiegman A., Hutten B., Groot E. et al.; Efficacy and safety of statin therapy im children with Familial Hypercholesterolemia. A randomized Controlled Trial; Pediatric Cardiology 2010:vol 5, No. 11, 1121-1126
22. Świątek H.; Ocena wpływu leczenia statynami na grubość kompleksu błona środkowa – śródbłonek u osób z hipercholesterolemią oraz prawidłowym ciśnieniem tętniczym; Rozprawka doktorska Gdańsk 2007








