Endokrynol. Ped. 11/2012;1(38):83-94
DOI: 10.18544/EP-01.11.01.1375
Wrodzona wielohormonalna niedoczynność przysadki – choroba wciąż późno rozpoznawana. Opis przypadków
1Klinika Pediatrii i Endokrynologii Warszawskiego Uniwersytetu Medycznego
2Studenckie Koło Naukowe Kliniki Pediatrii i Endokrynologii Warszawskiego Uniwersytetu Medycznego * Autorki miały taki sam udział w pracy
Słowa kluczowe: niedoczynność przysadki, hipoglikemia, dzieci
Streszczenie
Pomimo znacznego postępu wiedzy w ostatnich dekadach na temat genetycznego podłoża wielohormonalnej niedoczynności przysadki (WNP) i jej obrazu klinicznego nadal rozpoznanie nastręcza wielu trudności. W pracy przedstawiono przebieg kliniczny wrodzonej wielohormonalnej niedoczynności przysadki u trzech pacjentów. U każdego z nich objawy pojawiły się już w okresie noworodkowym, ale WNP rozpoznano dość późno. Różnorodny obraz wrodzonej WNP przysparza trudności diagnostycznych i opóźnia rozpoznanie. Epizody nawracającej hipoglikemii od okresu noworodkowego, przedłużająca się żółtaczka, micropenis, czy zanik nerwów wzrokowych powinny być zawsze rozważane jako objawy sugerujące wrodzoną WNP. Ze względu na zwiększone ryzyko obecności wad anatomicznych podwzgórza i przysadki u pacjentów z wrodzoną WNP należy u nich wykonywać rezonans magnetyczny jako badanie z wyboru, najlepiej obrazujące tę okolicę
Wstęp
Wielohormonalna niedoczynność przysadki (WNP) jest rozpoznawana w przypadku nieprawidłowej funkcji co najmniej dwóch osi hormonalnych zależnych od przysadki. Rozpoznanie wrodzonej WNP w wieku rozwojowym koreluje z wysokim ryzykiem obecności wady strukturalnej w okolicy podwzgórzowo-przysadkowej [1–8]. Z drugiej strony obecność wady strukturalnej w okolicy podwzgórzowo-przysadkowej zwiększa prawdopodobieństwo wystąpienia WNP [4]. Towarzyszy ona często takim wadom strukturalnym, jak agenezja ciała modzelowatego, dysplazja oczno-przegrodowa, holoprosencefalia, wrodzone torbiele pajęczynówki, może również występować u pacjentów z wadami linii pośrodkowej twarzy, jak rozszczep podniebienia. Najczęstszymi malformacjami związanymi z ryzykiem wrodzonej niedoczynności przysadki są: izolowana aplazja/hipoplazja przysadki, hipoplazja przedniego płata przysadki i/lub ektopia płata tylnego, zwłaszcza w połączeniu z agenezją szypuły przysadki, oraz zespół pustego siodła [9–13]. Przyczyną wrodzonej niedoczynności przysadki mogą być także urazy okołoporodowe, przy których można stwierdzić charakterystyczne zmiany w badaniu obrazowym OUN [12]. Jednocześnie niedoczynność przysadki u płodu zwiększa ryzyko przedłużonego czasu trwania ciąży oraz urazów okołoporodowych związanych z nieprawidłowym ułożeniem, porodami zabiegowymi [7, 9, 14]. U płodów z ułożeniem miednicowym w porównaniu z położeniem główkowym znacznie częściej spotyka się malformacje anatomiczne przysadki [12]. Położenie to najbardziej koreluje z ektopią płata tylnego przysadki, a dzieci najczęściej prezentują WNP [12]. U części pacjentów z wrodzoną WNP można potwierdzić genetyczne podłoże zaburzeń funkcji i budowy przysadki. Nieprawidłowości anatomiczne mają często związek z mutacjami genów odpowiedzialnych za jej rozwój. WNP z hipoplazją przedniego płata przysadki może być spowodowana mutacją genów: POU1F1, PROP1, HESX1, LHX3, LHX4, SOX 2 i SOX3 [15]. Hipoplazji może towarzyszyć ektopia tylnego płata przysadki, dzieje się tak w przypadku niektórych mutacji genów HESX 1, LHX4, SOX3 lub OTX2. Podłoże genetyczne WNP można stwierdzić także u pacjentów bez wad strukturalnych okolicy podwzgórzowo przysadkowej, przy niektórych mutacjach czynnika PROP1, LHX3 lub OTX2 [9]. W większości przypadków WNP i wady okolicy przysadkowo-podwzgórzowej występują sporadycznie, ale opisuje się także postaci rodzinne [16–19]. Uszkodzenia genu czynnika Pit-1 i Prop-1 odpowiadają za złożone niedobory dotyczące funkcji komórek tyreotropowych, somatotropowych i laktotropowych zarówno u zwierząt doświadczalnych, jak i u ludzi [20]. Pomimo znacznego postępu wiedzy w ostatnich dekadach na temat genetycznego podłoża wielohormonalnej niedoczynności przysadki i jej obrazu klinicznego nadal rozpoznanie nastręcza wielu trudności.
Celem pracy jest prezentacja przebiegu wrodzonej wielohormonalnej niedoczynności przysadki u trzech pacjentów, u których WNP rozpoznano dość późno, chociaż objawy sugerujące WNP były obecne od okresu noworodkowego.
Pacjent A.A. Pozostaje pod opieką naszej kliniki od 3 roku życia.
Chłopiec z ciąży I, urodzony w 42 tygodniu ciąży cięciem cesarskim z powodu przedłużającego się II okresu porodu. Oceniony na 9 punktów w skali Apgar. Masa ciała przy urodzeniu 3925 g, długość 54 cm. W badaniu przedmiotowym stwierdzono hipoplastyczne prącie (micropenis) i obustronne wnętrostwo. W pierwszej dobie życia pojawiły się zaburzenia oddychania i bezdechy. Rozpoznano ASD typu II. W badaniach dodatkowych stwierdzono hipoglikemię, obniżone stężenie lewotyroksyny: 0,56 ng/ml, przy TSH = 4,5 mIU/l. U chłopca rozpoznano pierwotną niedoczynność tarczycy, którą uznano za przyczynę hipoglikemii okresu noworodkowego, i rozpoczęto leczenie syntetyczną lewotyroksyną.
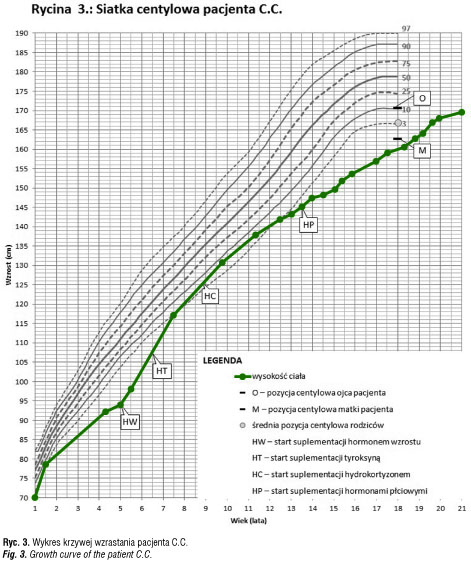
W okresie niemowlęcym i do 3 roku życia wielokrotnie modyfikowano dawkowanie i okresowo przerywano leczenie lewotyroksyną z powodu obniżania się TSH (<0,2mU/l) w momencie normalizacji stężenia fT4 i fT3. Chłopiec w okresie niemowlęcym wymagał rehabilitacji ze względu na obniżone napięcie mięśniowe i opóźniony rozwój psychoruchowy. Z powodu obustronnego wnętrostwa i hipoplastycznego prącia wykonano kariotyp: 46,XY. W drugim roku roku życia przebył jednostronną orchidopeksję. W okresie pooperacyjnym wystąpiła ciężka hipoglikemia, wymagająca infuzji 10% glukozy w dwóch pierwszych dobach po operacji. Krzywa wzrastania do trzeciego roku życia przebiegała tuż poniżej 3 centyla, przy średniej pozycji wysokości rodziców około 50 centyla (ryc. 1).
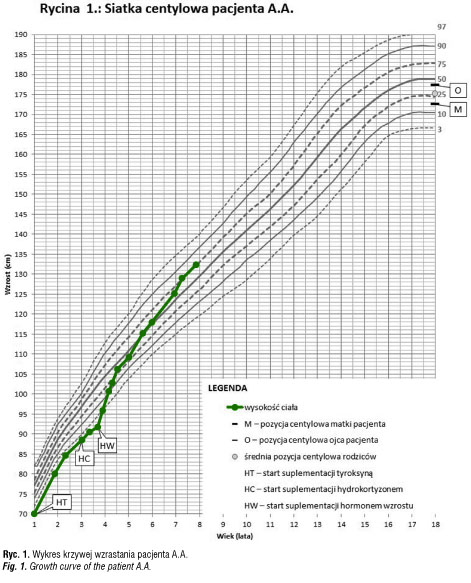
Przy przyjęciu do kliniki, w wieku 3 lat, wysokość ciała wynosiła 88 cm (<3c, -2,4 SDS), masa ciała 13,2 kg (10c; -0.9 SDS), BMI: 17,05 kg/m2.
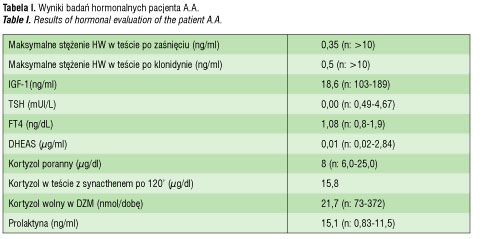
Stwierdzono niskie ciśnienie tętnicze: 80/50 mmHg, hipoplastyczne prącie o długości < 2cm (micropenis), lewostronne wnętrostwo, jądro prawe obecne w mosznie po sprowadzeniu operacyjnym. Rozwój psychoruchowy w zakresie dolnych wartości normy. Opóźniony rozwój mowy. Wiek szkieletowy nieharmonijny, między 1,5 a 2 rokiem życia. W badaniach laboratoryjnych stwierdzono niedokrwistość niedobarwliwą mikrocytarną. Glikemia na czczo: 71mg%. Wydalanie wolnego kortyzolu z moczem było obniżone, podobnie stężenie DHEAS i kortyzolu we krwi. Bardzo niskie było także stężenie IGF1. Wartości TSH i fT4 były typowe dla wtórnej niedoczynności tarczycy (tab. I). Rozpoznano wtórną niedoczynność nadnerczy i tarczycy. Po wprowadzeniu substytucji hormonalnej glikokortykoidami i tyroksyną przeprowadzono testy oceniające wyrzut hormonu wzrostu (tab. I).
U chłopca rozpoznano wielohormonalną niedoczynność przysadki w zakresie hormonu wzrostu, TSH, ACTH. Obustronne wnętrostwo i micropenis sugerowały, że u pacjenta wystąpił także niedobór gonadotropin. W badaniu MR stwierdzono hipoplastyczną część gruczołową przysadki położoną na dnie siodła tureckiego o wymiarach 5,7 x 4,6 x 6 mm, brak szypuły przysadki i ektopowy płat tylny zlokalizowany poniżej skrzyżowania nerwów wzrokowych. Po dołączeniu do substytucji preparatu hormonu wzrostu uzyskano dość szybką normalizację parametrów rozwoju fizycznego (ryc. 1).
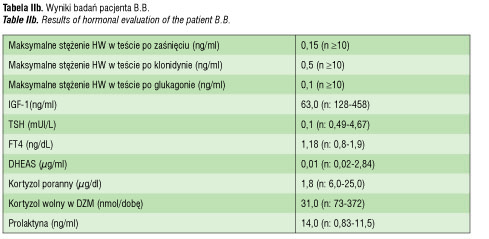
Pacjent B.B. Pozostaje pod opieką naszej kliniki od 11 roku życia.
Chłopiec urodzony z ciąży II, porodu II, w 41 tygodniu ciąży, siłami natury z pomocą ręczną. Masa urodzeniowa wynosiła 3450 g, długość ciała 54 cm. Był oceniony na 9 pkt w skali Apgar. W pierwszej dobie życia stwierdzono zaburzenia oddychania i krążenia oraz hipotermię. Podejrzewano
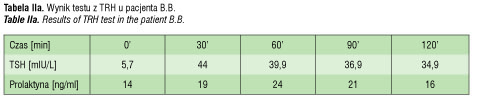
infekcję wewnątrzmaciczną i rozpoczęto antybiotykoterapię empiryczną po uprzednim pobraniu materiału do badań mikrobiologicznych. Nie udowodniono u chłopca etiologii infekcyjnej zaburzeń. W trakcie leczenia i nawadniania dożylnego płynami z dodatkiem 10% glukozy uzyskano poprawę stanu dziecka. Od okresu niemowlęcego chłopiec nie skupiał wzroku i nie wodził oczyma za przedmiotami. Stwierdzono znacznego stopnia niedowidzenie z zanikiem nerwów wzrokowych. W 9 miesiącu życia w tomografii komputerowej stwierdzono torbiel okolicy zbiornika okalającego i zbiornika skrzyżowania nerwów wzrokowych. Torbiel częściowo usunięto operacyjnie w 12 miesiącu życia. Utrzymywało się znacznego stopnia niedowidzenie. Krzywa wzrastania do 8 roku życia przebiegała 1–2 cm poniżej 3 centyla, podczas gdy średnia pozycja centylowa rodziców odpowiadała 50 centylowi (ryc. 2). W wieku 8 lat chłopiec ponownie przebył operację neurochirurgiczną z powodu znacznego odrostu torbieli. Z uwagi na brak możliwości bezpiecznego doszczętnego usunięcia torbieli wszczepiono zbiornik odbarczający Rickhama. W badaniu MR głowy uwidoczniono wówczas torbiel pajęczynówki uciskającą przysadkę. Szypuła przysadki nie była widoczna. Przeprowadzono diagnostykę funkcji przysadki: stwierdzono obniżone stężenie IGF1, bardzo niskie wartości hormonu wzrostu w testach stymulacyjnych. Wiek kostny był opóźniony o dwa lata. Znacznie obniżone było stężenie tyroksyny (0,49 ng/ml) przy nieznacznie podwyższonym TSH (5,7 mIU/L). Dodatkowo wykonano test z TRH, w którym odpowiedź TSH była nadmierna i przedłużona (tab. IIa). Rozpoznano wówczas pierwotną niedoczynność tarczycy i rozpoczęto leczenie substytucyjne lewotyroksyną w dawce docelowej 2 ug/kg/ dobę. Po wprowadzeniu leczenia pogorszyło się samopoczucie pacjenta, wystąpił brak apetytu, chłopiec był osłabiony, miał obniżone ciśnienie tętnicze, pojawiły się nudności i wymioty poranne. W kontrolnych badaniach laboratoryjnych stwierdzono obniżone stężenie TSH przy prawidłowej wartości fT4 i znacznie zredukowano dawkę lewotyroksyny (do 0,5ug/kg/dobę). Objawy ustąpiły, ale ponownie obniżyło się stężenie fT4. W kolejnych dwóch latach pacjent był leczony zmienną dawką tyroksyny od 0,5 do 2ug/kg masy ciała/ dobę. W tym czasie przyspieszył nieznacznie tempo wzrastania i osiągnął wysokość ciała na poziomie 10 centyla, jednak przy wyższej dawce tyroksyny wracały wcześniej opisane objawy.
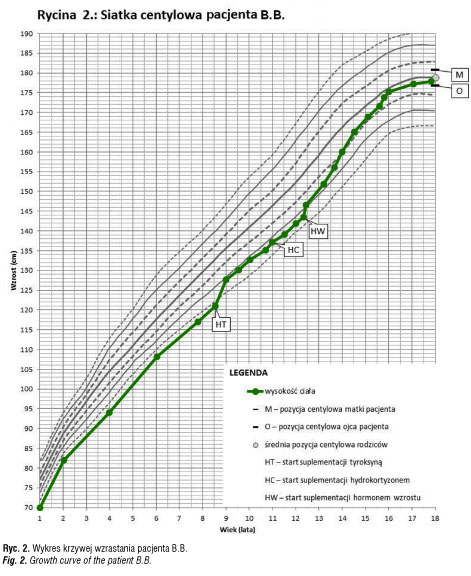
W 11 roku życia zgłosił się do naszej Kliniki. W badaniu przedmiotowym stwierdzono: wzrost 136,4 cm (10c; -1,2 SD), masę ciała 33,5 kg (>25c, -0,6 SD), BMI 18,0 (50-75c). Ciśnienie tętnicze wynosiło 94/76 mmHg. Stwierdzono niedowidzenie znacznego stopnia, skoliozę z rotacją w odcinku piersiowym i lędźwiowym, słabo rozwinięte mięśnie szkieletowe, nadmiar tkanki tłuszczowej rozmieszczony centralnie. Rozwój narządów płciowych w fazie dziecięcej, objętość jąder po 2ml. W badaniach dodatkowych: wiek kostny zgodny z metrykalnym; w badaniu USG – bardzo mała tarczyca, o objętości 1,6ml i prawidłowej echogeniczności; znacznie obniżone wydalanie wolnego kortyzolu z moczem, niskie ACTH, znacznie obniżone stężenie IGF1 (tab. IIb). Rozpoznano wtórną niedoczynność tarczycy i nadnerczy, zastosowano dodatkowo leczenie substytucyjne hydrokortyzonem, co spowodowało poprawę samopoczucia, normalizację ciśnienia tętniczego i ustąpienie objawów, które pojawiały się wcześniej przy dawkach lewotyroksyny zapewniających eutyreozę. Potwierdzono także niedobór hormonu wzrostu w testach stymulacyjnych z klonidyną oraz glukagonem i poza hydrokortyzonem i lewotyroksyną wprowadzono leczenie rekombinowanym hormonem wzrostu, uzyskując znaczną poprawę tempa wzrastania i prawidłową realizację przewidywanego wzrostu (ryc. 2).
Pacjent C.C. Pozostaje pod opieką naszej kliniki od 9 roku życia.
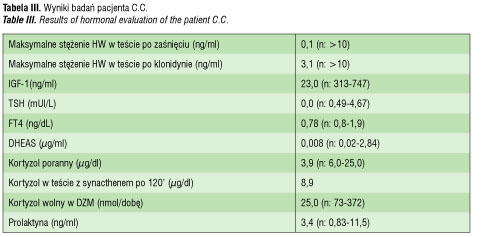
Chłopiec urodzony z ciąży II, porodu II, w 40 tygodniu ciąży, siłami natury. Masa urodzeniowa wynosiła 3800 g, długość 55 cm. W 5 minucie życia był oceniony na 9 pkt w skali Apgar. W pierwszej dobie życia wystąpiły drgawki i prężenia. Wykonano badanie EEG, które było prawidłowe, w badaniu neurologicznym nie stwierdzono istotnych nieprawidłowości. Od 2 miesiąca życia pacjent był wielokrotnie hospitalizowany w szpitalu rejonowym z powodu nawracających drgawek. W badaniach laboratoryjnych powtarzała się hipoglikemia i niedokrwistość niedobarwliwa. Od 4 roku życia chłopiec był leczony lekami przeciwpadaczkowymi bez efektu. W wieku 5 lat był diagnozowany z powodu hipoglikemii. W dobowym profilu glikemii minimalna wartość wynosiła 52 mg/dl w godzinach porannych, w teście głodowym występowało obniżenie glikemii do 60 mg/dl. Stwierdzono ponadto niedobór wysokości ciała (91cm, <3c) i opóźniony o 3 lata wiek kostny. Na podstawie wyników testów stymulacyjnych, w których wyrzut hormonu wzrostu był poniżej 1ng/ml, rozpoznano izolowany niedobór hormonu wzrostu. W TK głowy nie uwidoczniono nieprawidłowości. Pacjent został zakwalifikowany do leczenia hormonem wzrostu i od rozpoczęcia leczenia nie miał epizodów drgawek ani hipoglikemii. W 7 roku życia dołączono dodatkowo substytucję lewotyroksyną z uwagi na utrzymujący się obniżony poziom fT4, przy prawidłowym stężeniu TSH.
W wieku 9 lat chłopiec został przyjęty do naszej kliniki celem oceny wszystkich osi hormonalnych zależnych od przysadki. Rozpoznano wielohormonalną niedoczynność przysadki w zakresie hormonu wzrostu, ACTH i TSH (tab. III) i zastosowano pełne leczenie substytucyjne. Wykonano także rezonans magnetyczny OUN ze szczególnym uwzględnieniem przysadki, na którego podstawie rozpoznano zespół pustego siodła tureckiego. W wieku 14 lat u pacjenta rozpoznano dodatkowo hipogonadyzm hipogonadotropowy i rozpoczęto leczenie substytucyjne gonadotropiną kosmówkową, następnie testosteronem. Pacjent ma rozwój umysłowy w dolnej granicy normy. Zakończył edukację na poziomie trzeciej klasy gimnazjum.
Dyskusja
Obraz kliniczny wrodzonej WNP zależy od tego, jakie osie hormonalne i ile z nich funkcjonuje nieprawidłowo. U dzieci najczęstszą komponentą jest niedobór hormonu wzrostu [1, 9], który jako wada izolowana występuje z częstością od 1:4,000 do 1:8,000 żywo urodzonych noworodków [11, 21]. Wielohormonalna niedoczynność przysadki jest dużo rzadsza i występuje z częstością zbliżoną do 1:100,000 [22]. Wtórna niedoczynność tarczycy występuje jeszcze rzadziej, jej częstość szacuje się na 1:28,000–1:45,000 [23, 24]. W następnej kolejności wymienia się deficyt gonadotropin i ACTH, jakkolwiek według danych Zou i wsp. [10] w grupie 51 dzieci z WNP główną składową był niedobór hormonu wzrostu, w dalszej kolejności deficyt gonadotropin (35 z 51), a dopiero po nich niedobór TSH (31 z 51), ACTH (17 z 51) i prolaktyny (2 z 51). U opisanych przez nas pacjentów we wszystkich przypadkach potwierdzono niedobór hormonu wzrostu, wtórną niedoczynność tarczycy i nadnerczy, natomiast niedobór gonadotropin dotyczył tylko dwu pacjentów (tab. IV). U żadnego z nich nie stwierdzono niedoboru prolaktyny, choć u pacjenta z zespołem pustego siodła jej stężenie było stosunkowo niskie.
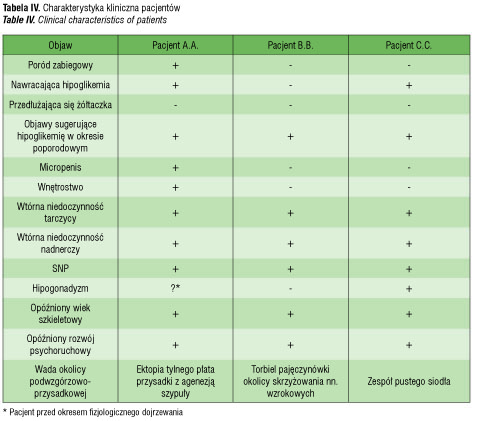
Zróżnicowany przebieg kliniczny WNP u pacjentów z deficytem tych samych hormonów przysadkowych sugeruje, że stopień niedoboru poszczególnych składowych zespołu też może być różny. Rozwój przedniego płata przysadki jest zależny od współdziałania szeregu czynników transkrypcyjnych, m.in. HESX1, PROP1, POU1F1, LHX3, LHX4, GLI2, SOX2 oraz SOX3 [9, 25, 26]. W zależności od rodzaju mutacji i od genu, którego dotyczy, fenotyp choroby jest inny. Defekty dotyczące genów biorących udział we wczesnej fazie rozwoju przysadki (np. LHX3, LHX4, HESX1) są przyczyną wielohormonalnej niedoczynności, której mogą towarzyszyć charakterystyczne wady anatomiczne, np. dysplazja przegrodowo-oczna. Przy uszkodzeniu genu odpowiedzialnego za końcowy etap rozwoju komórek produkujących jeden z hormonów efektem jest izolowany niedobór tego hormonu [9, 27]. Ponadto w zależności od tego, jaki czynnik transkrypcyjny jest uszkodzony, objawy kliniczne niedoboru hormonów mogą pojawić się w różnym okresie życia. W dysplazji przegrodowo-ocznej lub wadach związanych z mutacjami genu PROP1 zaburzenia hormonalne mogą być nieobecne przy urodzeniu i ujawniać się w późniejszych okresach życia [9, 26]. WNP często jest komponentą specyficznego zespołu objawów występujących przy mutacjach określonych genów, np. zespołu Pallistera–Halla (mutacja genu GLI3), zespołu Riegera (mutacja genu PITX2), może współistnieć z upośledzeniem umysłowym (mutacja genu SOX3) [28, 29], anophthalmią (mutacja genu SOX2) holoprosencefalią (mutacja genu GLI2), ograniczoną ruchomością szyi i tułowia (mutacja genu LHX3) [30, 31].
Najczęstszą genetyczną przyczyną WNP jest recesywna mutacja w genie PROP1, której częstość w postaciach rodzinnych sięga nawet 50% [32–34]. Wywołuje ona niedobór hormonu wzrostu, prolaktyny, TSH oraz gonadotropin i zwykle z rozwijającym się z opóźnieniem niedoborem ACTH [35–37]. Stopień niedoboru hormonów i wiek, w którym ujawniają się jego objawy, może być różny [9, 26, 35]. Obraz przysadki u tych pacjentów jest także zróżnicowany [9, 26]. Mutacja czynnika transkrypcyjnego POU1F1 skutkuje przede wszystkim niedoborem hormonu wzrostu i prolaktyny we wczesnym okresie życia oraz niedoborem TSH w późniejszym wieku [17]. Noworodki z wrodzoną WNP mogą nie prezentować żadnych objawów choroby. Szacuje się, że we wczesnym okresie noworodkowym objawy występują tylko u około 52% pacjentów z wrodzoną WNP [26]. Urodzeniowa długość i masa ciała są prawidłowe [38], jednak po urodzeniu zaznacza się słabszy przyrost masy ciała, a w pierwszych miesiącach życia wyraźne zahamowanie przyrostu długości ciała [9, 38]. Hipoglikemia należy do typowych objawów WNP u noworodka. Jest efektem niedoboru trzech istotnych osi hiperglikemizujących: hormonu wzrostu, glikokortykoidów oraz hormonów tarczycowych. Im cięższy niedobór, tym wcześniej ujawniają się objawy kliniczne. W okresie noworodkowym hipoglikemia może manifestować się zaburzeniami oddychania, niepokojem, drgawkami, które zwykle pojawiają się w pierwszych dniach, czasem nawet w pierwszych godzinach życia i mogą powracać. U naszych pacjentów objawy były niespecyficzne i jako ich przyczynę w pierwszej kolejności brano pod uwagę infekcję wewnątrzmaciczną lub wadę układu krążenia. U pacjentów z WNP częściej występuje przedłużająca się żółtaczka fizjologiczna, która głównie zależy od wtórnej niedoczynności tarczycy. Inne niespecyficzne objawy opisywane u pacjentów z wrodzoną WNP to hipotermia, sinica, asfiksja, hipotonia, objawy sugerujące niedrożność przewodu pokarmowego: wymioty, zaparcia [9, 39]. Niedobór TSH, ACTH i hormonu wzrostu może być odpowiedzialny za pojawienie się cholestazy, hepatosplenomegalię oraz wzrost GOT i GPT, którym typowo towarzyszy prawidłowy poziom GGTP [40–42]. Niezwykle rzadko w WNP rozwija się przełom nadnerczowy, w którym do wymienionych objawów dołączają się bradykardia, kwasica metaboliczna, hiponatremia, hiperkaliemia [43] lub tylko hiponatremia bez hiperkaliemii [9]. Niekiedy w przebiegu choroby może wystąpić hiperamonemia, która utrudnia dodatkowo różnicowanie, sugerując defekt metaboliczny [44]. Często pierwszym rozpoznaniem u dziecka z WNP jest niedoczynność tarczycy, jednak wybiórcza suplementacja T4 przyspiesza metabolizm innych hormonów, co zwiększa ryzyko nawracających ciężkich hipoglikemii [39]. U opisanych przez nas pacjentów A.A. i B.B. suplementacja syntetyczną tyroksyną spowodowała dekompensację dotychczasowej równowagi hormonalnej i ujawnienie się niedoczynności nadnerczy w postaci hipoglikemii i hipotonii ortostatycznej. Pacjent C.C. otrzymał substytucję tyroksyną już w trakcie leczenia hormonem wzrostu, którego działanie hiperglikemizujące zapobiegło nawrotom ciężkiej hipoglikemii.
W WNP niespecyficznym objawom ogólnym okresu noworodkowego mogą towarzyszyć zaburzenia rozwojowe oraz wady narządów płciowych. U chłopców bardzo istotnym i częstym objawem jest micropenis, któremu może towarzyszyć wnętrostwo [11, 39, 41]. Zaburzenia rozwoju oczu i linii pośrodkowej twarzoczaszki mogą występować pod postacią zeza, oczopląsu i rozszczepu wargi lub podniebienia [11, 44]. Rezonans magnetyczny OUN może ujawnić malformacje anatomiczne nie tylko okolicy przysadki, lecz także wiele innych nieprawidłowości, jak np. składowe dysplazji przegrodowo-ocznej: agenezja ciała modzelowatego, brak przegrody przezroczystej czy hipoplazja nerwu wzrokowego. Przykład pacjenta C.C., u którego zespół pustego siodła nie został opisany w tomografii komputerowej, pokazuje, że rezonans magnetyczny powinien być badaniem
z wyboru u pacjentów z dysfunkcją przysadki. U opisanych przez nas pacjentów w okresie noworodkowym występowały niespecyficzne objawy sugerujące hipoglikemię oraz towarzyszące czynniki ryzyka wrodzonej WNP: micropenis, wtórna niedoczynność tarczycy, hipoplazja nn wzrokowych, torbiel okolicy skrzyżowania nn. wzrokowych. Mimo to rozpoznanie ustalono z opóźnieniem. Według danych z literatury wrodzona WNP nadal jest rozpoznawana dość późno, rzadko w okresie noworodkowym lub niemowlęcym nawet przy wczesnej manifestacji klinicznej [35, 42, 44, 45]. Niedobór hormonów tarczycowych u dzieci przed trzecim rokiem życia i nawracające hipoglikemie mogą być przyczyną trwałegouszkodzenia OUN, dlatego tak ważne jest wczesne rozpoznanie i wczesne podjęcie leczenia, a także właściwa kolejność wprowadzanej substytucji. Według danych Browna [46] u dzieci z wrodzoną niedoczynnością przysadki stwierdzono upośledzenie funkcji kognitywnych, a średni iloraz inteligencji był istotnie niższy niż średnia populacyjna. U naszych pacjentów: 1. U A.A. stwierdzono opóźniony rozwój mowy, zaburzoną koordynację wzrokowo-ruchową, osiągnięcia szkolne na poziomie umożliwiającym realizację normalnego programu wymagają intensywnej dodatkowej pracy indywidualnej z dzieckiem. 2. Rozwój umysłowy pacjenta C.C. pozostaje w dolnej granicy normy. 3. U pacjenta B.B. nie stwierdza się upośledzenia rozwoju umysłowego.
Podsumowanie
Różnorodny obraz kliniczny wrodzonej wielohormonalnej niedoczynności przysadki przysparza trudności diagnostycznych i opóźnia rozpoznanie. W pracy opisujemy pacjentów, u których rozpoznanie można było postawić znacznie wcześniej. Chcemy zwrócić uwagę na konieczność oceny wszystkich osi hormonalnych u pacjentów z niedoczynnością przysadki i na właściwą interpretację wyników badań oceniających funkcjonowanie osi sprzężeń zwrotnych. Przykład naszych pacjentów pokazuje, że najczęściej stwarza to trudności w przypadku wtórnej niedoczynności tarczycy. Epizody nawracającej hipoglikemii od okresu noworodkowego, przedłużająca się żółtaczka lub micropenis powinny być zawsze rozważane jako objawy sugerujące wrodzoną WNP. W związku z tym, że u pacjentów z wrodzoną WNP zwykle stwierdza się zmiany anatomiczne w okolicy podwzgórzowo- przysadkowej, należy wykonywać rezonans magnetyczny jako badanie z wyboru, najlepiej obrazujące tę okolicę.
Piśmiennictwo
1. Dutta P., Bhansali A., Singh P. et al.; Clinico-radiological correlation in childhood hypopituitarism; Indian. Pediatr. 2010:47, 615-618
2. Ochi M., Morikawa M., Yoshimoto M. et al.; Growth retardation due to idiopathic growth hormone deficiencies: MR findings in 24 patients; Paediatr. Radiol. 1992:22, 477-480
3. Arends N. J. T., Lip W. V.D., Robben S. G. F. et al.; MRI findings of the pituitary gland in short children born small for gestational age (SGA) in comparison with growth hormone-deficient (GHD) children and children with normal stature; Clin. Endocrinol. (Oxf) 2002:57, 719-724
4. Bozzola M., Mengarda F., Sartirana P. et al.; Long-term follow-up evaluation of magnetic resonance imaging in the prognosis of permanent GH deficiency; Eur. J. Endocrinol. 2000:143(4), 493-496
5. Cacciari E., Zucchini S., Carlà G. et al.; Endocrine function and morphological findings in patients with disorders of the hypothalamo-pituitary area: a study with magnetic resonance; Arch. Dis. Child. 1990:65, 1199-1202
6. Mehta A., Hindmarsh P.C., Mehta H. et al.; Congenital hypopituitarism: clinical, molecular and neuroradiological correlates; Clin. Endocrinol. 2009:71(3), 376-382
7. Maghnie M., Larizza D., Triulzi F. et al.; Hypopituitarism and stalk agenesis: A congenital syndrome worsened by breech delivery?; Horm. Res 1991:35 (3-4), 104-108
8. Kucharska A., Bołtuć A., Chojnacki J.; Występowanie zmian strukturalnych w okolicy podwzgórzowo-przysadkowej i ich znaczenie kliniczne u pacjentów z somatotropinową niedoczynnością przysadki; Pediatric Endocrinology, Diabetes and Metabolism 2010:16 (3), 142-147
9. Alatzoglou K.S., Dattani M.T.; Genetic forms of hypopituitarism and their manifestation in the neonatal period; Early Hum. Dev. 2009:85, 705-712
10. Zou C.C., Chen X., Liang L. et al.; Childhood combined pituitary hormone deficiency: A five-year retrospective study; Endocrinologist 2010:20 (3), 125-127
11. Lammoglia J.J., Eyzaguirre F., Unanimous N. et al.; Congenital hypopituitarism: report of 23 cases; Rev. Med. Chil. 2008:136, 996-1006
12. Fujita K., Matsuo N., Mori O.; The association of hypopituitarism with small pituitary, invisible stalk, type II Arnold Chiari malformations and syringomyelia in seven patients born in brech position: a further proof of birth injury theory on the pathogenesis of idiopathic hypopituitarism; Eur. J. Pediatr. 1992:151, 266-270
13. Vanelli S., Avataneo T., Benso I. et al.; Magnetic resonance and the diagnosis of short stature of hypothalamic-hypophyseal origin; Acta Paediatr. Scand. Suppl. 1993:82,155-161
14. Coya R., Vela A., Perez de Nanclares G. et al.; Panhypopituitarism: Genetic versus acquired etiological factors; J. Pediatr. Endocrinol. Metab. 2007:20 (1), 27-36
15. Parks J.S., Brown M.R., Hurley D.L. et al.; Heritable disorders of pituitary development; J. Clin. Endocrinol. Metab. 1999:84, 4362-4370
16. Wu W., Cogan J.D., Pfaffle R.W. et al.; 1X Mutations in PROP1 cause familial combined pituitary hormone deficiency; Nat. Genetics 1999:18, 147-149
17. Pfäffle R.W., DiMattia G.E., Parks J.S. et al.; Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia; Science 1992:257, 1118-1121
18. Pfäffle R.W., Blakenstein O., Wüller S., Kentrup H.; Combined pituitary hormone deficiency: Role of Pit-1 and Prop-1; Acta Paediatr. 1999:(Suppl 433), 33-41
19. Reynaud R., Chadli-Chaieb M., Vallette-Kasic S.; A familial form of congenital hypopituitarism due to a PROP1 mutation in a large kindred: phenotypic and in vitro functional studies; J. Clin. Endocrinol. Metab. 2004:89(11), 5779-5786
20. Cheng T.C., Beamer W.G., Phillips III J.A. et al.; Etiology of growth hormone deficiency in Little, Ames and Snell dwarf mice; Endocrinology 1983:113,1669-1678
21. Webb E.A., Dattani M.T.; Understanding hypopituitarism; Paediatrics and Child Health 2011:21(7), 289-294
22. Fischer D.A.; Second International Conference on Neonatal Thyroid Screening: progress report; J. Pediatr. 1983:102(5), 653-654
23. Borck G., Topaloglu A.K., Korsch E. et al.; Four New Cases of Congenital Secondary Hypothyroidism due to a Splice Site Mutation in the Thyrotropin- Gene: Phenotypic Variability and Founder Effect; J. Clin. Endocrinol. Metab. 2004:89(8), 4136-4141
24. Hanna C.E., Krainz PL, Skeels MR et al.; Detection of congenital hypopituitary hypothyroidism: ten-year experience in The Northwest Regional Screening Program; J. Pediatr. 1986:109, 959-964
25. Mullis P.E.; Transcription Factors in Pituitary Gland Development and Their Clinical Impact on Phenotype; Horm. Res. 2000:54, 107-119
26. Mehta A., Dattani M.T.; Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism; Clin. Endocrinol. (Oxf) 2008:22, 191-206
27. Dattani M.T.; Growth hormone deficiency and combined pituitary hormone deficiency: Does the genotype matter?; Clin. Endocrinol. (Oxf) 2005:63 (2), 121-130
28. Solomon N.M., Nouri S., Warne G.L. et al.; Increased gene dosage at Xq26-q27 is associated with X-linked hypopituitarism; Genomics 2002:79(4), 553-559
29. Laumonnier F., Ronce N., Hamel B.C. et al.; Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency; Am. J. Hum. Genet. 2002:71(6), 1450-1455
30. Pfaeffle R.W., Savage J.J., Hunter C.S. et al.; Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation; J. Clin. Endocrinol. Metab. 2007:92, 1909-1919
31. Netchine I., Sobrier M.L., Krude H. et al.; Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency; Nat. Genet. 2000:25, 182-186
32. Turton J.P., Mehta A., Raza J. et al.; Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD); Clin. Endocrinol. (Oxf) 2005:63, 10-18
33. Vieira T.C., Boldarine V.T., Abucham J.; Molecular analysis of PROP1, PIT1, HESX1, LHX3, and LHX4 shows high frequency of PROP1 mutations in patients with familial forms of combined pituitary hormone deficiency; Arq. Bras. Endocrinol. Metabol. 2007:51, 1097-1103
34. Cogan J.D., Wu W., Phillips III J.A. et al.; The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency; J. Clin. Endocrinol. Metab. 1998:83, 3346-3349
35. Flück C., Deladoey J., Rutishauser K. et al.; Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting in the substitution of Arg3Cys AT codon 120 (R120C); J. Clin. Endocrinol. Metab. 1998:83, 3727-3734
36. Asteria C., Oliveira J.H., Abucham J. et al.; Central hypocortisolism as part of combined pituitary hormone deficiency due to mutations of PROP-1 gene; Eur. J. Endocrinol. 2000:143(3), 347-352
37. Agarwal G., Bhatia V., Cook S. et al.; Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion; J. Clin. Endocrinol. Metab. 2000:85(12), 4556-4561
38. Mehta A., Hindmarsh P.C., Stanhope R.G. et al.; The role of growth hormone in determining birth size and early postnatal growth, using congenital growth hormone deficiency (GHD) as a model; Clin. Endocrinol. (Oxf) 2005:63, 223-231
39. Salisbury D.M., Leonard J.V., Dezateux C.A. et al.; Micropenis: an important early sign of congenital hypopituitarism; Br. Med. J. (Clin. Res. Ed.) 1984:288, 621-622
40. Sheehan A.G., Martin S.R., Stephure D. et al.; Neonatal cholestasis, hypoglycemia, and congenital hypopituitarism; J. Pediatr. Gastroenterol. Nutr. 1992:14, 426-430
41. Binder G., Martin D.D., Kanther I. et al.; The course of neonatal cholestasis in congenital combined pituitary hormone deficiency; J. Pediatr. Endocrinol. Metab. 2007:20 (6), 695-701
42. Karnsakul W., Sawathiparnich P., Nimkarn S. et al.; ; Ann. Hepatol. 2007:6 (2), 97-103
43. Lee W.S., Lum L.C.S., Harun F.; Addisonian-like crisis in congenital hypothyroidism and cholestatic jaundice; Med. J. Malaysia 2003:58 (2), 279-281
44. Bhoyar A., Short A.; Congenital Hypopituitarism Associated with Hyperammonemia; Indian J. Pediatr. 2009:76 (3), 327-328
45. Nagasaki K., Ohashi T., Hiura M. et al.; Neonatal identification of congenital hypopituitarism with an invisible pituitary stalk and pituitary aplasia: Usefulness of early growth hormone replacement; Clin. Pediatr. Endocrinol. 2005:14 (SUPPL. 24),93-96
46. Brown K., Rodgers J., Johnstone H. et al.; Abnormal cognitive function in treated congenital hypopituitarism; Arch. Dis. Child. 2004:89, 827-830








