Endokrynol. Ped. 12/2013;2(43):57-68
DOI: 10.18544/EP-01.12.02.1451
Profil chorób autoimmunologicznych u pacjentów z zespołem Turnera
Katedra i Klinika Pediatrii, Endokrynologii i Diabetologii Dziecięcej Śląskiego Uniwersytetu Medycznego w Katowicach
Słowa kluczowe: zespół Turnera, choroby autoimmunologiczne, chromosom X
Streszczenie
Zespół Turnera jest spowodowany liczbowymi i/lub strukturalnymi nieprawidłowościami w obrębie chromosomu X. Rozpoznawany z częstością 1 na 2000-2500 żywo urodzonych noworodków płci żeńskiej manifestuje się klinicznie niedoborem wzrostu, brakiem dojrzewania płciowego, płetwiastą szyją, puklerzowatą klatką piersiową i obrzękiem limfatycznym. W ostatnich latach zwraca się uwagę na jego związek z chorobami autoimmunologicznymi. Poza najczęściej rozpoznawanym zapaleniem tarczycy typu Hashimoto wśród częstych wymienić należy: cukrzycę typu 1, celiakię, choroby zapalne jelit i szereg jednostek dermatologicznych. Szeroko rozpowszechniona jest obecność autoprzeciwciał. Przyczyna tej szczególnej podatności jest trudna do określenia, niemniej wśród czynników predysponujących mogą uczestniczyć genetyczne (w tym związane z chromosomem X i autosomami) i hormonalne. Aneuploidia, haploinsuficjencja i pochodzenie chromosomu X, podobnie jak konkretne geny, a wśród nich FOXP3 i PTPN22 mogą partycypować w patogenezie. Terapia rekombinowanym hormonem wzrostu, jak i rola estrogenów, androgenów oraz zaburzenia w syntezie glikokortykoidów, nie powinny pozostać niezauważone, odkąd znana jest wzajemna interferencja układu dokrewnego i odpornościowego. Zespół Turnera reprezentuje szereg zaburzeń w profilu immunologicznym, w tym nieprawidłowy rozkład subpopulacji limfocytów i ich wzmożoną apoptozą, które również zostały omówione. Autorzy opracowania starają się całościowo przedstawić złożoną problematykę autoagresji w zespole Turnera, opisując dotychczasowy stan wiedzy
Wstęp
Zespół Turnera (ZT) może być rozpoznany jedynie u dziewczynki lub kobiety, u których jednocześnie stwierdza się charakterystyczne cechy fenotypowe oraz znamienny dla zespołu kariotyp z obecnym tylko jednym prawidłowych chromosomem X [1]. Po raz pierwszy klasyczne stygmaty zespołu zostały prawdopodobnie opisane w XVIII wieku. Jego nazwa pochodzi jednak od nazwiska amerykańskiego internisty Henry Turnera, który w roku 1938 zaprezentował siedem pacjentek z charakterystycznymi i powtarzającymi się cechami: niskim wzrostem, brakiem cech dojrzewania, płetwiastością szyi i koślawością łokci [2]. Dwadzieścia lat później, w roku 1959, specyficzne współistnienie tych cech zostało powiązane przez Forda z liczbowymi lub strukturalnymi aberracjami chromosomu X [3]. Częstość występowania zespołu szacuje się na 1 na 2000–2500 żywo urodzonych noworodków płci żeńskiej [4]. W aspekcie bieżących wskaźników urodzeń w Polsce można przypuszczać, że co roku rodzi się około 80–100 dziewcząt z ZT. Z uwagi na profil dolegliwości, wśród których na czoło wysuwa się niskorosłość oraz objawy związane z dysgenezją gonad, lekarzem koordynującym proces diagnostyczny i terapeutyczny jest z reguły endokrynolog. W ostatnim okresie zwraca się dość dużą uwagę na wyraźnie zwiększone ryzyko chorób autoimmunologicznych w tej grupie chorych.
W opracowaniu przedstawiamy w oparciu o dane literatury oraz doświadczenia własne aktualne poglądy odnoszące się do rodzaju i patogenezy zaburzeń autoimmunologicznych u dziewcząt i kobiet z ZT.
Częstość chorób autoimmunologicznych w zespole Turnera
Ryzyko chorób autoimmunologicznych u pacjentek z zespołem Turnera (ZT) jest około dwukrotnie wyższe w stosunku do ogólnej populacji żeńskiej i czterokrotnie wyższe w odniesieniu do chorób dominujących u płci męskiej (np. cukrzyca typu 1) [5]. ZT predysponuje do autoimmunologicznego zapalenia tarczycy, celiakii, cukrzycy typu 1, bielactwa, łysienia plackowatego, wrzodziejącego zapalenia jelita grubego oraz choroby Crohna [6–11].
Z chorób o podłożu autoimmunologicznym na pierwszym miejscu pod względem częstości w ZT znajdują się choroby tarczycy [8]. Przeciwciała przeciwtarczycowe stwierdza się u ponad 20% chorych z ZT [9, 10, 12–15] Częstość chorób autoimmunologicznych tarczycy wzrasta dość wyraźnie po 13 r.ż. [12], a szczyt zachorowań przypada na trzecią dekadę [13]. Według niektórych autorów nie należy spodziewać się przeciwciał przeciwtarczycowych i wynikających z tego niedoczynności tarczycy w przebiegu zapalenia przed 8 rokiem życia [9, 14, 15], choć w naszych ostatnich badaniach najmłodsza pacjentka z ZT, u której stwierdzono ich obecność, miała 5,5 roku [12]. W grupie dziewcząt z ZT niedoczynność tarczycy, najczęściej w przebiegu jej zapalenia i bez objawów klinicznych hipotyreozy, można stwierdzić u co czwartej pacjentki [9, 12]. Na podstawie długofalowych badań wykazano, że wśród kobiet z ZT częstość niedoczynności tarczycy, najczęściej w przebiegu limfocytarnego zapalenia, wzrasta rocznie o dalsze 3,2% [14].
Dane co do częstości choroby Gravesa-Basedowa czy cukrzycy typu 1 w ZT są sprzeczne. Bakalov i wsp. [16] nie potwierdzają ich zwiększonego ryzyka wśród chorych z ZT, ale jednocześnie przyznają, że może to wynikać z nie dość dużej grupy chorych objętej badaniem.
Częstość celiakii w ZT ocenia się na około 4–6% [1, 17]. Według Arslan [18] i Bonamico [19] ryzyko jej wystąpienia jest 11-krotnie wyższe niż w ogólnej populacji, ale stosunek pacjentów objawowych do tych bez stygmatów klinicznych choroby jest porównywalny.
Ryzyko wystąpienia choroby Leśniowskiego-Crohna czy wrzodziejącego zapalenia jelita grubego wzrasta 2–10-krotnie w porównaniu do statystyk w ogólnej populacji [8]. Według Larizza i wsp. [20] ta pierwsza ma znacznie gorsze rokowanie u pacjentek z ZT: ujawnia się w młodszym wieku i istotnie częściej kończy się interwencją chirurgiczną. Z dermatologicznych jednostek chorobowych występujących częściej w ZT niż ogólnej populacji należy wymienić: łuszczycę (2 razy częściej niż w ogólnej populacji) [21], łysienie plackowate (3x częściej) [22] oraz bielactwo (u 2,7% pacjentek z ZT) [23].
Wśród innych częściej niż w ogólnej populacji występujących w ZT chorób o podłożu autoimmunologiczny należy wymienić młodzieńcze zapalenie stawów (6-krotnie wyższe ryzyko zachorowania) [24], chorobę Addisona oraz pierwotną żółciową marskość wątroby.
Poszukiwanie przyczyn zwiększonej częstości chorób autoimmunologicznych w zespole Turnera
Patogeneza większości chorób autoimmunologicznych rozpatrywana jest obecnie wieloczynnikowo, z nakładającym się wpływem czynników
genetycznych i środowiskowych. Brak istotnie różnego narażenia środowiskowego powoduje, że większą część ryzyka przypisujemy czynnikom wewnętrznym.
Przyczyny, które predysponują do autoimmunogenności w grupie pacjentek z ZT, nie są do końca poznane, niemniej poszukuje się zaburzeń w układzie immunologicznym, uwzględnia się udział czynników genetycznych związanych przede wszystkim z nieprawidłowościami w obrębie chromosomu X oraz wpływ zaburzeń hormonalnych i stosowanej terapii hormonalnej. Zwiększone ryzyko chorób autoimmunologicznych najczęściej jednak próbuje tłumaczyć się haploinsuficjencją genów chromosomu X [25], pochodzeniem chromosomu X, nadmierną produkcją cytokin prozapalnych [26], hipogonadyzmem oraz koniecznością zastosowania estrogenoterapii.
Zaburzenia immunologiczne
Poza częstszym występowaniem chorób autoimmunologicznych u pacjentek z ZT wiele prac potwierdza również niedobory w zakresie odporności komórkowej jak i humoralnej [27–35]. Skłania to do poszukiwania zaburzeń immunologicznych w ZT na różnych etapach funkcjonowania układu immunologicznego. Niestety większość przeprowadzonych w tym kierunku analiz opiera się na wynikach uzyskanych na mało licznych grupach, często bardzo różniących się wiekiem czy rodzajem kariotypu.
Standardowe badania morfologii krwi obwodowej w ZT, poza podwyższoną liczbą trombocytów, nie ujawniły odchyleń od normy [36]. Wyniki badań grupy z Karolinska University Hospital, przeprowadzonych głównie w celu poszukiwania przyczyn istotnie częściej występujących w ZT zapaleń ucha środkowego i podejrzenia w związku z tym niedoborów odporności, wskazują, że ogólne stężenia immunoglobulin klas głównych IgA, IgM, IgD, IgG i podklas IgG1, IgG2, IgG3 oraz IgG4 oscylują w granicach normy [37]. Niemniej uwzględniając wcześniejsze doniesienia, których wyniki nie były tak jednoznaczne [27–29, 38], nie można uznać ich za rozstrzygające [37]. Również ilość komórek NK, makrofagów i aktywność hemolityczna dopełniacza nie wydają się zmienione u chorych z ZT [37, 39].
Dokładniejsza analiza subpopulacji leukocytów wykazała natomiast różnice w stosunku do populacji ogólnej [40,41]. Uznawanym faktem jest obniżenie stosunku limfocytów T CD3+CD4+/CD3+CD8+, a także zmniejszenie odpowiedzi na mitogen [37, 39, 40, 42, 43]. Wśród autorów nie ma zgodności, czy wynika to ze spadku odsetka komórek CD3+CD4+ [40], czy wzrostu odsetka CD3+CD8+ [37, 39]. Zaburzenia w głównych liniach limfocytów T w ZT mogą być spowodowane ich większą podatnością na apoptozę. Gupta i wsp. [41] obserwowali w ZT wzrost spontanicznej apoptozy limfocytów CD4+ i CD8+ związanej ze wzrostem ekspresji CD95L (FasL). Receptor Fas (CD95) należy do nadrodziny TNF indukującej apoptozę i zawierającej domenę śmierci, która aktywuje dwa niezależne i przeciwstawne szlaki w komórce. Jeden z nich prowadzi właśnie do apoptozy poprzez aktywację FADD i kaspazy 8. Chociaż ekspresja Fas na komórkach CD4+ i CD8+ u osób z ZT jest podobna jak w populacji zdrowej, jest prawdopodobne, że w ZT występują dodatkowe nieprawidłowe sygnały aktywujące Fas. Ponadto autorzy ci wykazali, że ekspresja antyapoptycznych białek FLIP i IAP (inhibitory of apoptosis protein) w limfocytach ZT jest obniżona odpowiednio 50- i 3-krotnie. Być może zwiększona apoptoza subpopulacji limfocytów odgrywa główną rolę w niedoborach odporności towarzyszących ZT.
Zaburzenia stanu równowagi między reakcją odpornościową a mechanizmami odpowiedzialnymi za wyciszenie stanu zapalnego mogą prowadzić do chorób autoimmunologicznych. Zaangażowane są tutaj m.in. limfocyty naiwne T CD4+, które w obecności odpowiednich cytokin i czynników transkrypcyjnych różnicują się do kilku subpopulacji. Ich zachwiana ekspresja mogłaby tłumaczyć odmienności w stosunku do zdrowej populacji, czego jednak badania w grupie z ZT nie potwierdziły [40,43]. Wśród komórek CD4+ wyróżniamy m. in. komórki Th1, Th2, Th17 i Treg. Te pierwsze, zdolne do wytwarzania interferonu gamma i IL2, promują odpowiedź komórkową, prozapalną. Th2 produkując IL4, IL5, IL10 IL13 związane są z odpowiedzią humoralną, kojarzy się je także z atopią. Th17, biorące udział w odpowiedzi prozapalnej, przeciwbakteryjnej i przeciwgrzybicznej, produkują IL17,21,22. I wreszcie powstająca w obecności IL10 i TGFβ - populacja komórek regulatorowych o fenotypie CD4+CD25+FOXP3+. Treg, odkryte w połowie lat 90. ubiegłego wieku przez Sakaguchi i wsp., mają zasadniczy udział w indukowaniu tolerancji organizmu na własne antygeny, Są one zdolne do ingerencji w proces autoimmunizacji przez hamowanie komórek autoreaktywnych w bezpośrednim kontakcie i poprzez produkcję TGFβ o działaniu immunosupresyjnym [40,43]. Fan i wsp. [40] potwierdzili w grupie pacjentek z ZT istotny wzrost ilości limfocytów Th17 i jednoczesny wzrost liczby Treg w stosunku do zdrowych kobiet z grupy kontrolnej. Mogłoby to sugerować przyrost wyrównawczy limfocytów T regulatorowych przy jednoczesnej niewydolności funkcjonalnej, choć eksperymentalnie wykluczono taką możliwość [39, 40]. W szeregu komórek B (CD19+) nie zanotowano zmian ilościowych, w przeciwieństwie do istotnie większej liczby głównych producentów przeciwciał – komórek plazmatycznych (CD19-CD138+) [40].
Miano wielu specyficznych autoprzeciwciał w ZT jest podwyższone. Na czoło wysuwają się przeciwciała p/tyreoperoksydazowe
i p/tyreoglobulinowe oraz przeciw komórkom okładzinowym żołądka [42]. Nie odnosi się to jednak do autoprzeciwciał związanych z APS (autoimmune polyendocrine syndrome) czy chorobą Addisona (przeciwciała przeciw 17alfa-hydroksylazie, dekarboksylazie L-aminokwasów aromatycznych, hydroksylazie tyrozynowej i tryptofanowej oraz 21-hydroksylazie), co potwierdziły badania Stenberg i wsp. [44].
Czynniki genetyczne
Predyspozycja do rozwoju chorób autoimmunologicznych nie jest cechą unikalną zespołu Turnera. Sama aneuploidia predysponuję do wzrostu autoimmunogenności, czego przykładem jest zwiększone ryzyko chorób autoimmunologicznych w zespole Downa i Klinefeltera. Podobnie jak w przypadku zwiększonego ryzyka chorób autoimmunizacyjnych, również w etiopatogenezie objawów niedoboru odporności rozpatruje się wpływ ilości materiału chromosomu X (kobiety 47,XXX oraz mężczyźni 47,XXY mają wyższe stężenia immunoglobulin) [45] oraz negatywne oddziaływanie niedoboru estrogenów [28].
a. Czynniki związane z chromosomem X
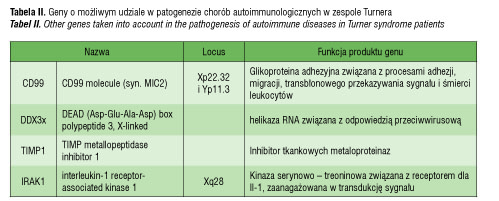
Według Pessach [46] i Bianchi [47] jest co najmniej 9 genów zlokalizowanych na chromosomie X, które mogą mieć wpływ regulujący na funkcje immunologiczne, a których mutacje mogą prowadzić do niedoborów immunologiczncych i zaburzeń autotolerancji (tabela I). Do nich między innymi należy gen FOXP3 (forkhead box P3, Xp11.23-q13.3), kodujący czynnik transkrypcyjny istotny dla prawidłowej funkcji regulatorowych limfocytów T (limfocytów CD4+CD25+), oddziaływający z NFAT (nuclear factor of activated T cells) [46, 48]. Badania na myszach jednoznacznie potwierdziły, że obniżona ekspresja tego genu zaburza funkcję limfocytów T regulatorowych i przyczynia się do wystąpienia chorób autoimmunologicznych [49]. Co więcej, udowodniono, że mutacja FOXP3 jest odpowiedzialna za wystąpienie zespołu IPEX (immunodysregulation, polyendocrinopathy and enteropathy, X-linked syndrome), na który składają się wcześnie ujawniająca się i o ciężkim przebiegu enteropatia, cukrzyca typu I, zapalenie skóry, liczne przeciwciała w surowicy krwi oraz anemia hemolityczna, małopłytkowość, nefropatia czy zapalenie tarczycy [48]. Wyniki obserwacji co do znaczenia FOXP3 w patogenezie chorób autoimmunologicznych w ZT nie potwierdziły, by liczba jego prawidłowych kopii przekładała się na ilość czy funkcję supresorową limfocytów T regulatorowych in vitro [39]. Należy jednak pamiętać, że również w IPEX liczba limfocytów T, granulocytów, stężenie immunoglobulin i aktywność dopełniacza nie wykazują odchyleń, przy czym występują liczne autoprzeciwciała [39, 49]. Potrzebne są dalsze badania, by wyjaśnić tę kwestię. Inne geny, których udział rozważa się w patogenezie chorób autoimmunologicznych w zespole Turnera, prezentuje tabela II.
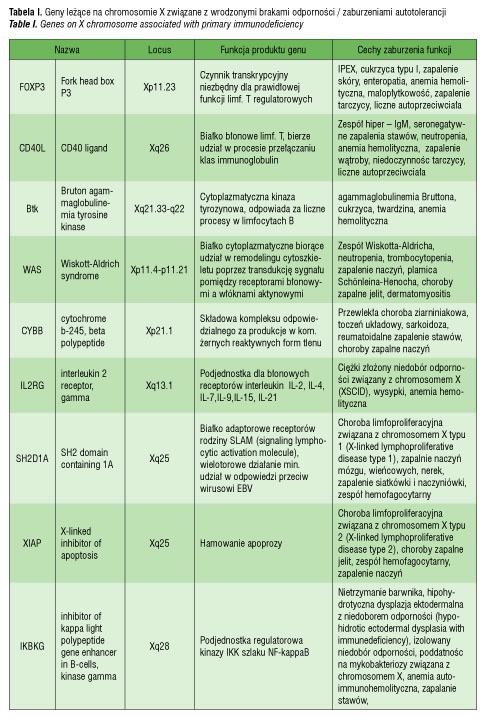
Powszechnie znany jest fakt częstszego występowania chorób autoimmunologicznych u płci żeńskiej. W eksperymentalnych modelach zwierzęcych potwierdzono, że większe ryzyko autoimmunogenności u samic może wynikać z różnic hormonalnych: estrogeny mają wpływ nasilający, a androgeny hamujący aktywność chorób z autoagresji [50, 51]. Nie udało się jednak tak ścisłej zależności potwierdzić u ludzi. Według Chitnis i wsp. [52] czynnikiem mogącym wpływać na wzrost autoimmunogenności jest proces inaktywacji chromosomu X, w którym jeden z chromosomów X ulega inaktywacji lub transkrypcyjnemu wyciszeniu we wczesnych etapach rozwoju embrionalnego. W normalnych warunkach efektem tego jest mozaicyzm tkankowy, w którym w jednych komórkach ekspresji ulega chromosom X pochodzenia matczynego, a w drugich pochodzenia ojcowskiego. Proces inaktywacji chromosomu X u płci żeńskiej naraża na sytuację, w której autoantygeny z nim związane mogą uniknąć prezentacji w grasicy lub tkankach obwodowych zaangażowanych w proces indukcji tolerancji.
Obecność tylko jednego prawidłowego chromosomu X, w kontekście procesu inaktywacji, może tłumaczyć istotnie większe ryzyko występowania chorób autoimmunologicznych nie tylko w odniesieniu do mężczyzn, ale i kobiet. Co prawda Chitnis i wsp. nie znaleźli na to jednoznacznych dowodów, ale uzyskane wyniki nie pozwoliły również na odrzucenie ich hipotezy [52].
Sugerowano, że częstsze występowanie chorób autoimmunologicznych w ZT może wiązać się z matczynym pochodzeniem chromosomu X oraz obecnością materiału pochodzącego z chromosomu Y [5, 53]. Jednak z drugiej strony wyniki badań zarówno Bakalov i wsp. [16], jak i Sagi i wsp. [54] nie potwierdzają, by obecność chromosomu X pochodzenia matczynego czy ojcowskiego korelowała z wyższym ryzykiem ujawnienia się autoimmunologii.
Biorąc pod uwagę wyniki prezentowane przez grupę Bakalov i wsp. [16], można wnioskować, że haploinsuficjencja pewnych genów chromosomu X odgrywa dominującą rolę w patogenezie choroby Hashimoto i innych zaburzeń o podłożu autoimmunologicznym.
W związku z brakiem lub zaburzeniami strukturalnymi chromosomu X będącymi podłożem molekularnym zespołu Turnera oczywista wydaje sie sugestia, że zwiększone prawdopodobieństwo zjawisk autoimmunizacyjnych wynika z nieprawidłowej ilości kopii genów zlokalizowanych na tym chromosomie. Przesłanką potwierdzającą niniejszą tezę może być obserwacja porównywalnego ryzyka zachorowania na SLE wśród pacjentów z zespołem Klinefeltera (XXY) i zdrowych kobiet (XX), istotnie wyższego od zdrowych mężczyzn (XY) [55].
Wyższą skłonność do schorzeń autoimmunologicznych obserwowano w przypadku obecności izochromosomu X z ramion długich (iXq) [5, 56]. Mechanizm, który wyjaśniałby, dlaczego monosomia w zakresie ramienia krótkiego chromosomu X wiąże się ze zwiększoną częstością schorzeń autoimmunologicznych, nie został poznany. Niektórzy spekulują, że może to wynikać z haploinsuficjencji FOXP3 oraz obecności jednocześnie trisomii w zakresie ramienia długiego, która nasila prozapalną i apoptotyczną odpowiedź [16]. Chociaż według niektórych obecność iXq wiąże się z 12-krotnie wyższym ryzykiem zachorowania na wrzodziejące zapalenie jelit [5, 11], to w badaniu grupy z NIH nie uzyskano tak istotnie zwiększonego ryzyka zapaleń jelit (a jedynie tendencję), natomiast potwierdzono silny związek tego kariotypu z limfocytarnym zapaleniem tarczycy [16]. W wypadku izochromosomu występują trzy kopie genów obecnych na długim ramieniu i tylko jedna na krótkim, tak więc nadmiar któregoś z genów ramienia długiego lub nierównowaga pomiędzy ramionami mogłaby być priorytetowa dla rozwoju limfocytarnego zapalenia tarczycy. Mortensen i wsp. [57] badający częstość występowania różnych autoprzeciwciał w ZT z iXq potwierdzają ją jedynie w odniesieniu do obecności przeciwciał anty-GAD-65 (ang. anti-glutamic-aciddecarboxylase 65). Podobną zależność między kariotypem z iXq i przeciwciałami anty-GAD-65, a także cukrzycą typu 2 obserwowali Bakalov i wsp. [58], ale jednocześnie nie potwierdzali zwiększonej częstości cukrzycy typu 1 (<0,5%). Według nich sama haploinsuficjencja genu(ów) ramienia krótkiego chromosomu X zaburza funkcję komórek β wysp trzustkowych. Ponadto „nadmiar” materiału genetycznego wynikający z obecności iXq może ten stan pogłębiać przez zmianę (ekspresji) innych genów istotnych w prawidłowym rozwoju, funkcji i przeżyciu komórek β oraz przez stymulację przewlekłej autoimmunizacji uszkadzającej te komórki [57]. Wcześniejsze dane o 11-krotnie wyższym ryzyku wystąpienia cukrzycy typu 1 w populacji ZT [7] nie znajdują potwierdzenia i z pewnością wymagają weryfikacji. Z drugiej strony istotnie częstsze występowanie przeciwciała anty-GAD-65 w ZT w porównaniu z ogólną populacją (4% vs. 1,1%) budzi większą czujność i powinno mobilizować do regularnych badań kontrolnych.
W oparciu o wyniki tak popularnych ostatnio analiz mikromacierzy potwierdzono również przesunięcia ekspresji genów w stronę parametrów zapalnych, w tym obniżenie ekspresji obecnego na długim ramieniu X, TSC22D3 („przeciwzapalny czynnik transkrypcyjny” związany z działaniem glikokortykosteroidów), wzrost ekspresji CRP (C Reactive Protein), oraz zmiany w zaanagażowanych w apoptozę transkryptów XIAP, MAP3K1, MAPK8 [41,58].
b. Czynniki związane z autosomami
Rodzeństwo chorych z zespołem Turnera o zrównoważonym kariotypie prezentuje zwiększone ryzyko wystąpienia chorób autoimmunizacyjnych. Ponadto zastanawiające jest, że pacjentki mają większe prawdopodobieństwo wystąpienia autoprzeciwciał, gdy takie występują u ojca, a już takiego związku nie ma, gdy występują u matki [42]. Nawiązując w tym miejscu do ryzyka związanego z chromosomem X, jego pochodzenie, matczyne bądź ojcowskie, nie ma związku, przynajmniej w odniesieniu do obecności przeciwciał przeciwtarczycowych, dlatego sugerowane niegdyś teorie o różnicach w imprintingu genomowym nie wydają się przekonujące, odkąd wiadomo, że chromosom X nie podlega imprintingowi [59–61]. Część zagrożenia brakiem autotolerancji może być związana z genami autosomalnymi, co tłumaczyłoby zwiększoną zapadalność wśród rodzeństwa, jednak klasycznie wiązane z rozwojem chorób autoimmunologicznych allele głównego układu zgodności tkankowej MHC klasy II (chromosom 6) nie wykazują szczególnego sprzężenia z ZT. Oczywiście obecność serotypów wiązanych z autoimmunologią, DQ2 czy DQ8 determinuje większe prawdopodobieństwo celiakii również i w tej populacji [1, 42]. Związek taki występuje z allelami klasy I, HLA-A31 i HLA-B38 [20]. Jak sugerowano, ta frapująca segregacja ma prawdopodobnie powiązanie z immunologicznymi mechanizmami ochronnymi dla płodu, u którego są obecne powyższe izoformy, co determinuje wyższą masą urodzeniową. Serotypy, które promują przeżycie płodów (większość z zespołem Turnera ulega poronieniu), jednocześnie wiązałyby się z występowaniem autoagresji, co znalazło potwierdzenie dla allelu A31 [62], choć ten sam allel chroni przed rozwojem cukrzycy typu I resztę populacji [63].
Ostatnie badania dowiodły, że poza układem MHC w rozwoju chorób autoimmunologicznych istotną rolę odgrywa polimorfizm pojedynczego nukleotydu (SNP – single-nucleotide polymorphism) genu PTPN22 (1p13.3–13.1). PTPN22 koduje fosfatazę tyrozynową LYP hamującą transdukcję sygnału po pobudzeniu receptora TCR (T-cell receptor). Nie ma zgodności co do tego, czy polimorfizm C1858T jest mutacją typu wzrost czy utraty funkcji, niemniej klinicznie objawia się jako cecha dominująca. Z nie do końca ustalonych przyczyn predysponuje do rozwoju m.in. ziarniniakowatości Wegenera, choroby Hashimoto, SLE i reumatoidalnego zapalenia stawów. Jego istotnie wyższą obecność notowano wśród brazylijskiej populacji chorych z zespołem Turnera. Ze względu na znaczne zróżnicowanie dystrybucji allelu wśród różnych populacji oraz niepewną rolę, jaką odgrywa dla przeżycia, znaczenie tej obserwacji trudno na razie oceniać [64–66].
Udział hormonów (estrogeny, androgeny, glikokortykosteroidy, rekombinowany hormon wzrostu)
W komórkach immunokompetentnych stwierdzono obecność receptorów dla estrogenów [67, 68], androgenów [67], hormonu wzrostu [69] i glikokortykosteroidów [70]. Te proste dowody na współdziałanie układu immunologicznego i endokrynnego (w istocie wydzielonych sztucznie) skłaniają do rozważenia wpływu hormonów na obecność autoagresji w zespole Turnera. Jest to o tyle istotne, że terapia w ZT zakłada substytucję hormonów płciowych oraz leczenie rekombinowanym hormonem wzrostu. Dokładny wpływ hormonów na układ immunologiczny jest złożony i opisanie go znacznie przekracza ramy niniejszego opracowania, dlatego zostanie zarysowany w niezbędnym skrócie.
Choć wiadomo, że estrogeny wpływają na układ immunologiczny między innymi przez wpływ na rozwój limfocytów T [71] i hamowanie procesu zapalnego [72], to trudno jednoznacznie określić ich udział w rozwoju chorób autoimmunologicznych [73,74]. W wysokich stężeniach stymulują komórki T CD4+ do produkcji IL-4, IL-10 i IFN-γ oraz hamują produkcję TNF. Wskazuje to na działanie obniżające autoimmunogenność limfocytów T.
W tych samych stężeniach estradiol pobudza sekrecję przeciwciał przez komórki B (CD5+), ale jednocześnie hamuje szpik w zakresie prekursorów limfocytów B. Tak więc estrogeny mogą nasilać autoimmunogenność komórek B, ale też obniżać autoimmunogenność limfocytów T [72].
Uważa się, że hormony płciowe mogą istotnie przyczyniać się do zwiększenia ryzyka choroby autoimmunologicznej przez kształtowanie odpowiedzi Th1/Th2. W przypadku chorób autoimmunologicznych, w których patogenezie odgrywa rolę przede wszystkim odpowiedź Th1, obserwuje się złagodzenie objawów w okresach zwiększenia stężenia estrogenów nasilających odpowiedź Th2. Większość chorób autoimmunologicznych zdecydowanie częściej występuje u kobiet – zapalenie tarczycy, SLE czy twardzina układowa odpowiednio 18, 9 i 12-razy częściej niż w populacji mężczyzn. W okresie ciąży, w zależności od jednostki chorobowej, może dochodzić do osłabienia (jak np. w stwardnieniu rozsianym, reumatoidalnym zapaleniu stawów) bądź zaostrzenia (jak w SLE) jej objawów [63, 74].
Obecność receptorów estrogenowych (ER) stwierdzono w większości leukocytów ludzi zdrowych z wyłączeniem komórek CD4+ i makrofagów [67,68].Czy ma to jednak przełożenie na obraz kliniczny zespołu Turnera? Nic na to nie wskazuje.
Czas oraz natężenie ekspozycji na estrogeny nie wydaje się wpływać na zapadalność na choroby autoimmunologiczne [12,16]. Ponadto terapia estrogenami nie ma wpływu na ekspresję czynników transkrypcyjnych RORγT, T-bet, GATA3 w limfocytach oraz nie zmienia stosunku subpopulacji komórek immunokompetentnych [42].
Androgeny z kolei wykazują zdecydowanie efekt hamujący na układ immunologiczny [72].
Hipogonadyzm jest istotnym czynnikiem predysponującym do wystąpienia chorób autoimmunologicznych tarczycy. Potwierdziły to pierwsze prospektywne badania grupy z NIH: częstość przewlekłego zapalenia tarczycy w grupie chorych z pierwotną niedoczynnością jajników oszacowano na 15% i była ona istotnie wyższa w porównaniu z populacją kobiet amerykańskich (5,8%) [16]. Wykazano że ryzyko rozwoju choroby autoimmunologicznej rośnie w porządku: zdrowi mężczyźni, zdrowe kobiety, chore z pierwotną niewydolnością jajników, chore z zespołem Turnera. W tejże kolejności maleje jednocześnie stężenie androgenów. Dlatego istotą może nie być niekorzystne działanie estrogenów, ale brak ochronnej (immunosupresyjnej) roli androgenów. Ekspresję receptora androgenowego wykazują komórki B i makrofagi. Chociaż brakuje dokładnych analiz, to ta odwrotna korelacja jest zastanawiająca. Nie można wykluczyć, że
niewydolność jajników może prowadzić do niedoboru całkiem innej substancji o działaniu supresyjnym (np. metabolity steroidów) [16, 67]. Ponadto pamiętając, że receptor androgenowy kodowany jest przez gen długiego ramienia chromosomu X (Xq12), ważne wydaje się uwzględnienie wrażliwości leukocytów na androgeny.
Immunomodulacja glikokortykosteroidów odbywa sie na wielu poziomach: przez wpływ na komórki dendrytyczne, makrofagi, neutrofile, limfocyty B i T. Hormony te mają udział zarówno w grasiczym rozwoju limfocytów T, jak i późniejszej obwodowej tolerancji, m.in. przez wpływ na apoptozę oraz syntezę cytokin. Defekty w ich syntezie (nie tylko nadnerczowej, ale i miejscowej) mogą prowadzić do przetrwania autoreaktywnych form limfocytów [70]. Interesujące, że wśród pacjentek z zespołem Turnera oraz wśród pacjentów z zespołem Klinefeltera wykrywa się znacząco wysoki odsetek heterozygot genu CYP21 (Val281Leu), co przekłada się na zaburzenie steroidogenezy. Autorzy opisywanej pracy sugerują, że posiadanie owej mutacji może promować przeżycie płodu przez interferencję steroidów w zjawiska immunologiczne obecne w czasie ciąży [75]. Niedobór 21-hydroksylazy (CYP21) związany jest z zaburzeniami immunologicznymi, spośród których na czoło wysuwa sie obniżenie odsetka limfocytow T regulatorowych i obecność autoprzeciwciał [76]. Tak więc już częściowy niedobór tego enzymu przy uwzględnieniu innych aspektów mógłby się przekładać na zwiększone ryzyko choroby autoimmunologicznej.
Optymalna terapia pacjentek z zespołem Turnera zakłada wieloletnie podawanie rekombinowanego hormonu wzrostu (GH). Obecność receptorów dla GH stwierdzono na powierzchni limfocytów T, B, NK, monocytów oraz w licznych narządach limfatycznych, gdzie odbywa sie ponadto produkcja GH in situ. Nie potwierdzono wpływu terapii GH na stężenie immunoglobulin ani ilość różnych podtypów limfocytów [38]. Wśród jego licznych działań wymienić należy natomiast przyspieszenie tymopoezy, powstawania limfocytów B i nasilenie produkcji przeciwciał, co w rezultacie mogłoby prowadzić do niezrównoważenia układu immunologicznego [69]. Pomimo to nie stwierdzono związku pomiędzy rozwojem klinicznie jawnych chorób a terapią GH. Niektórzy [77] badacze [78] wskazują na zwiększony odsetek występowania przeciwciał przeciwtarczycowych [20]. Podobnie analiza mikromacierzy leukocytów pobranych od pacjentek z ZT poddanych terapii GH nie wykazała zmian ekspresji ich genów w odpowiedzi na hormon, chociaż w wypadku pacjentów z niedoborem hormonu wzrostu (GHD) regulacji podlegały m.in. SOCS1 (suppressor of cytokine signalling) czy TNFAIP3 (tumor necrosis factor alpha – induced protein-3) [79]. Warto w tym miejscu wspomnieć, że obecność przeciwciał przeciwnarządowych jest związana z gorszą ostateczną odpowiedzią na leczenie GH [80].
Z innych czynników ryzyka autoimmunologii (głównie tarczycy) sugeruje się palenie tytoniu, niemniej w badaniu Bakalov i wsp. nie wiązało się to ze zwiększonym ryzykiem wystąpienia choroby Hashimoto u pacjentek z ZT [16].
Choć nie udało do chwili obecnej wyjaśnić patomechanizmu zwiększonej częstości chorób autoimmunologicznych w tej grupie chorych, to już od dawna wiadomo o pewnych zaburzeniach w funkcjonowaniu układu odpornościowego [81]. U pacjentek z ZT stwierdza się niedobory odporności zarówno komórkowej, jak i humoralnej [40]. Trzy dekady temu pojawiły się pierwsze doniesienia o związku ZT z obniżoną ilością krążących w limfocytach T i B [28,29]. Ponadto wykazano obniżony stosunek CD4/CD8, słabą chemotaksję komórek polimorfonuklernych [38], obniżone stężenie immunoglobulin w podklasie IgG i IgM [27], zwiększoną ilość naturalnych komórek cytotoksycznych (NK, CD16+) oraz upośledzoną odpowiedź komórek T na mitogeny [29]. W ostatnio opublikowanych badaniach stwierdzono, że stężenie prozapalnych cytokin IL6 i TGFβ1 jest istotnie wyższe, a cytokin przeciwzapalnych TGFβ2 i IL10 niższe w grupie chorych z ZT zarówno w porównaniu do chorych z niewydolnością jajników, jak i do grupy kontrolnej zdrowych kobiet [16,82].
Jak widać, nie ma zgodności co do etiopatogenezy zwiększonego ryzyka występowania chorób autoimmunologicznych w grupie chorych z potwierdzonym zespołem Turnera. Tylko nieliczne z cytowanych prac opierały się na dostatecznie dużych populacjach. Temat pozostaje otwarty i wymaga dalszych badań klinicznych.
Piśmiennictwo
1. Bondy C.A.; Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group; J. Clin. Endocrinol. Metab. 2007:92, 10-25
2. Turner H.H.; A syndrome of infantilism, congenital webbed neck, and cubitus valgus; Endocrinology 1938:23, 566-574
3. Ford C.E., Jones K.W., Polani P.E. et al.; A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner’s syndrome); Lancet 1959:1, 711-713
4. Hook E.B., Warburton D.; The distribution of chromosomal genotypes associated with Turner’s syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism; Hum. Genet. 1983:64, 24-27
5. Jørgensen K.T., Rostgaard K., Bache I. et al.; Autoimmune diseases in women with Turner’s syndrome; Arthritis Rheum. 2010:62, 658-666
6. Lowenstein E.J., Kim K.H., Glick S.A.; Turner’s syndrome in dermatology; J. Am. Acad. Dermatol. 2004:50, 767-776
7. Elsheikh M., Dunger D.B., Conway G.S. et al.; Turner’s syndrome in adulthood; Endocr. Rev. 2002:23, 120-140
8. Gravholt C.H., Juul S., Naeraa R.W. et al.; Morbidity in Turner syndrome; J. Clin. Epidemiol. 1998:51, 147-158
9. Livadas S., Xekouki P., Fouka F. et al.; Prevalence of thyroid dysfunction in Turner’s syndrome: a long-term follow-up study and brief literature review; Thyroid 2005:15, 1061-1066
10. Elsheikh M., Wass J.A., Conway G.S.; Autoimmune thyroid syndrome in women with Turner’s syndrome--the association with karyotype; Clin. Endocrinol. (Oxf) 2001:55, 223-226
11. Hayward P.A., Satsangi J., Jewell D.P.; Inflammatory bowel disease and the X chromosome; QJM 1996:89, 713-718
12. Gawlik A., Gawlik T., Januszek-Trzciakowska A. et al.; Incidence and dynamics of thyroid dysfunction and thyroid autoimmunity in girls with Turner’s syndrome: a long-term follow-up study; Horm. Res. Paediatr. 2011:76, 314-320
13. Chiovato L., Larizza D., Bendinelli G. et al.; Autoimmune hypothyroidism and hyperthyroidism in patients with Turner’s syndrome; Eur. J. Endocrinol. 1996:134, 568-575
14. El-Mansoury M., Bryman I., Berntorp K. et al.; Hypothyroidism is common in turner syndrome: results of a five-year follow-up; J. Clin. Endocrinol. Metab. 2005:90, 2131-2135
15. Germain E.L., Plotnick L.P.; Age-related anti-thyroid antibodies and thyroid abnormalities in Turner syndrome; Acta. Paediatr. Scand. 1986:75, 750-755
16. Bakalov V.K., Gutin L., Cheng C.M. et al.; Autoimmune disorders in women with turner syndrome and women with karyotypically normal primary ovarian insufficiency; J. Autoimmun. 2012:38, 315-321
17. Frost A.R., Band M.M., Conway G.S.; Serological screening for coeliac disease in adults with Turner’s syndrome: prevalence and clinical significance of endomysium antibody positivity; Eur. J. Endocrinol. 2009:160, 675-679
18. Arslan N., Esen I., Demircioglu F. et al.; The changing face of celiac disease: a girl with obesity and celiac disease; J. Paediatr. Child. Health. 2009:45, 317-318
19. Bonamico M., Pasquino A.M., Mariani P. et al.; Prevalence and clinical picture of celiac disease in Turner syndrome; J. Clin. Endocrinol. Metab. 2002:87, 5495-5498
20. Larizza D., Calcaterra V., Martinetti M.; Autoimmune stigmata in Turner syndrome: when lacks an X chromosome; J. Autoimmun. 2009:33, 25-30
21. Watabe H., Kawakami T., Kimura S. et al.; Childhood psoriasis associated with Turner Syndrome; J. Dermatol. 2006:33, 896-898
22. Tebbe B., Gollnick H., Müller R. et al.; Alopecia areata and diffuse hypotrichosis associated with Ullrich-Turner syndrome. Presentation of 4 patients; Hautarzt 1993:44, 647-652
23. Brazzelli V., Larizza D., Martinetti M. et al.; Halo nevus, rather than vitiligo, is a typical dermatologic finding of turner’s syndrome: clinical, genetic, and immunogenetic study in 72 patients; J. Am. Acad. Dermatol. 2004:51, 354-358
24. Zulian F., Schumacher H.R., Calore A. et al.; Juvenile arthritis in Turner’s syndrome: a multicenter study; Clin. Exp. Rheumatol. 1998:16, 489-494
25. Invernizzi P., Miozzo M., Selmi C. et al.; X chromosome monosomy: a common mechanism for autoimmune diseases; J. Immunol. 2005:175, 575-578
26. Gravholt C.H., Hjerrild B.E., Mosekilde L. et al.; Body composition is distinctly altered in Turner syndrome: relations to glucose metabolism, circulating adipokines, and endothelial adhesion molecules; Eur. J. Endocrinol. 2006:155, 583-592
27. Jensen K., Petersen P.H., Nielsen L.E. et al.; Serum immunoglobulin M, G and A concentration levels in Turner’s syndrome compared with normal women and men; Hum. Gen. 1976:31, 329-334
28. Lorini R., Ugazio A.G., Cammareri V. et al.; Immunoglobulin levels, T-cell markers, mitogens responsiveness and thymic hormone activity in Turner’s syndrome; Thymus 1983:5, 61-66
29. Cacciari E., Masi M., Fantini M.P. et al.; Serum immunoglobulins and lymphocyte subpopulation derangement in Turner’s syndrome; J. Immunogenet 1981:8, 337-344
30. Al-Attas R.A., Rahi A.H.S., Ahmed E.L. Fe.; Common variable immunodeficiency with CD4+ T lymphopenia and over production of IL-2 receptor associated with Turner’s syndrome and dorsal kyphoscoliosis; J. Clin. Pathol. 1997:50, 876-879
31. Robson S.C., Potter P.C.; Common variable immunodeficiency associated with Turner’s syndrome; J. Clin. Lab. Immunol. 1990:32, 337-344
32. Hochberg Z., Aviram M., Rubin D. et al.; Decreased sensitivity of insulin-like growth factor I in Turner’s syndrome a study of monocytes and T lymphocytes; Eur. J. Clin. Invest. 1997:27, 543-547
33. Kurabayashi T., Yasuda M., Fujimaki T. et al.; Effect of hormone replacement therapy on spinal cord bone density and T lymphocyte subsets in premature ovarian failure and Turner’s syndrome; Int. J. Gynaecol. Obstet. 1993:42, 25-31
34. Mock M.U.R., Vogelsang H., Jager L.; Selective T cell deficiency in Turner’s syndrome; J. Invest. Allerg. Clin. Immunol. 2000:5, 312-313
35. Shewior S., Brand M., Santer R.; Celiac disease and selective IgA deficiency in a girl with atypical Turner’s syndrome; J. Ped. Gastroent. Nutr. 1999:28, 353-354
36. Jez W., Irzyniec T.; The values of peripheral blood cell count parameters in women with Turner’s syndrome; Pol. Merkur. Lekarski 2010:29, 247-249
37. Stenberg A.E., Sylvén L., Magnusson C.G. et al.; Immunological parameters in girls with Turner syndrome; J. Negat. Results Biomed. 2004:3, 6
38. Rongen-Westerlaken C., Rijkers G.T., Scholtens E. et al.; Immunologic studies Turner’s syndrome before and during treatment with growth hormone; J. Pediatr. 1991:119, 268-272
39. Su M.A., Stenerson M., Liu W. et al.; The role of X-linked FOXP3 in the autoimmune susceptibility of Turner Syndrome patients; Clin. Immunol. 2009:131, 139-144
40. Fan H., Wang D., Zhu H. et al.; Lymphocyte subpopulations in Chinese women with Turner syndrome; Arch. Gynecol. Obstet. 2012:285, 749-755
41. Gupta S., Chiplunkar S., Gupta A. et al.:; Increased spontaneous, tumor necrosis factor receptor- and CD95 (Fas)-mediated apoptosis in cord blood T-cell subsets from Turner’s syndrome; Genes. Immun. 2003:4, 239-243
42. Larizza D., Martinetti M., Lorini R. et al.; Parental segregation of autoimmunity in patients with Turner’s syndrome: preferential paternal transmission?; J. Autoimmun. 1999:12, 65-72
43. Eisenstein E.M., Williams C.B.; The T(reg)/Th17 cell balance: a new paradigm for autoimmunity; Pediatr. Res. 2009:65, 26R-31R
44. Stenberg A.E., Sylvén L., Hedstrand H. et al.; Absence of autoantibodies connected to autoimmune polyendocrine syndrome type I and II and Addison’s disease in girls and women with Turner syndrome; J. Negat. Results Biomed. 2007:6, 10
45. Rhodes K., Markham R.L., Maxwell P.M. et al.; Immunloglobulins and the X-chromosome.; Br. Med. J. 1969:3,439-441
46. Pessach I.M., Notarangelo L.D.; X-linked primary immunodeficiencies as a bridge to better understanding X-chromosome related autoimmunity; J. Autoimmun. 2009:33, 17-24
47. Bianchi I., Lleo A., Gershwin M.E. et al.; The X chromosome and immune associated genes; J. Autoimmun. 2012:38, J187-192
48. van der Vliet H.J., Nieuwenhuis E.E.; IPEX as a result of mutations in FOXP3.; Clin. Dev. Immunol. 2007:2007, 89017
49. Salomon B., Lenschow D.J., Rhee L. et al.; B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes; Immunity 2000:12, 431-440
50. Cooper G.S., Dooley M.A., Treadwell E.L. et al.; Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus; Arthritis Rheum. 1998:41, 1714-1724
51. Cutolo M., Sulli A., Villaggio B. et al.; Relations between steroid hormones and cytokines in rheumatoid arthritis and systemic lupus erythematosus; Ann. Rheum. Dis. 1998:57, 573-577
52. Chitnis S., Monteiro J., Glass D. et al.; The role of X-chromosome inactivation in female predisposition to autoimmunity; Arthritis Res. 2000:2, 399-406
53. Uematsu A., Yorifuji T., Muroi J. et al.; Parental origin of normal X chromosomes in Turner syndrome patients with various karyotypes: implications for the mechanism leading to generation of a 45,X karyotype; Am. J. Med. Genet. 2002:111, 134-139
54. Sagi L., Zuckerman-Levin N., Gawlik A. et al.; Clinical significance of the parental origin of the X chromosome in turner syndrome.; J. Clin. Endocrinol. Metab. 2007:92, 846-852
55. Cooney C.M., Bruner G.R., Aberle T. et al.; 46,X,del(X)(q13) Turner’s syndrome women with systemic lupus erythematosus in a pedigree multiplex for SLE; Genes. Immun. 2009:10, 478-481
56. Sparkes R.S., Motulsky A.G.; Hashimoto’s disease in Turner’s syndrome with isochromosome X; Lancet 1963:1, 947
57. Mortensen K.H., Cleemann L., Hjerrild B.E. et al.; Increased prevalence of autoimmunity in Turner syndrome--influence of age; Clin. Exp. Immunol. 2009:156, 205-210
58. Bakalov V.K., Cheng C., Zhou J. et al.; X-chromosome gene dosage and the risk of diabetes in Turner syndrome; J. Clin. Endocrinol. Metab. 2009:94, 3289-3296
59. Mathur A., Stekol L., Schatz D. et al.; The parental origin of the single X chromosome in Turner syndrome: lack of correlation with parental age or clinical phenotype; Am. J. Hum. Genet. 1991:48, 682-686
60. Chu C.E., Connor J.M.; Molecular biology of Turner’s syndrome; Arch. Dis. Child. 1995:72, 285-286
61. Sharp A.J., Stathaki E., Migliavacca E. et al.; DNA methylation profiles of human active and inactive X chromosomes; Genome. Res. 2011:21, 1592-1600
62. Larizza D., Martinetti Bianchi M., Lorini R. et al.; Autoimmunity, HLA, Gm and Km polymorphisms in Turner’s syndrome; Autoimmunity 1989:4, 69-78
63. Amur S., Parekh A., Mummaneni P.; Sex differences and genomics in autoimmune diseases; J. Autoimmun. 2012:38, J254-265
64. Bottini N., Musumeci L., Alonso A. et al.; A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes; Nat. Genet. 2004:36, 337e8
65. Bianco B., Verreschi I.T., Oliveira K.C. et al.; PTPN22 polymorphism is related to autoimmune disease risk in patients with Turner syndrome; Scand. J. Immunol. 2010:72, 256-259
66. Burn G.L., Svensson L., Sanchez-Blanco C. et al.; Why is PTPN22 a good candidate susceptibility gene for autoimmune disease?; FEBS Lett. 2011:585, 3689-3698
67. Rubtsov A.V., Rubtsova K., Kappler J.W. et al.; Genetic and hormonal factors in female-biased autoimmunity; Autoimmun. Rev. 2010:9, 494-498
68. Cunningham M., Gilkeson G.; Estrogen receptors in immunity and autoimmunity; Clin. Rev. Allergy. Immunol. 2011:40, 66-73
69. Hattori N.; Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system; Growth. Horm. IGF Res. 2009:19, 187-197
70. Baschant U., Tuckermann J.; The role of the glucocorticoid receptor in inflammation and immunity; J. Steroid. Biochem. Mol. Biol. 2010:120, 69-75
71. Pernis A.B.; Estrogen and CD4+ T cells; Curr. Opin. Rheumatol 2007:19, 414-420
72. Straub R.H.; The complex role of estrogens in inflammation; Endocr. Rev. 2007:28, 521-574
73. Trolle C., Hjerrild B., Cleemann L. et al.; Sex hormone replacement in Turner syndrome; Endocrine. 2012:41, 200-219
74. Rubtsov A.V., Rubtsova K., Kappler J.W. et al.; Genetic and hormonal factors in female-biased autoimmunity; Autoimmun. Rev. 2010:9, 494-498
75. Mantovani V., Dondi E., Larizza D. et al.; Do reduced levels of steroid 21-hydroxylase confer a survival advantage in fetuses affected by sex chromosome aberrations?; Eur. J. Hum. Genet. 2002:10, 137-140
76. Parlato F., Pisano G., Brillante M. et al.; Immunological pattern in patients with 21-hydroxylase deficiency; J. Endocrinol. Invest. 1994:17, 635-639
77. Nienhuis H.E., Rongen-Westerlaken C., Geertzen H.G. et al.; Long-term effect of human growth hormone therapy on the prevalence of autoantibodies in Turner syndrome. The Dutch Growth Hormone Working Group; Horm. Res. 1993:39, 49-53
78. Ivarsson S.A., Ericsson U.B., Nilsson K.O., Gustafsson J. et al.; Thyroid autoantibodies, Turner’s syndrome and growth hormone therapy; Acta. Paediatr. 1995:84, 63-65
79. Whatmore A.J., Patel L., Clayton P.E.; A pilot study to evaluate gene expression profiles in peripheral blood mononuclear cells (PBMCs) from children with GH deficiency and Turner syndrome in response to GH treatment; Clin. Endocrinol. (Oxf) 2009:70, 429-434
80. Bettendorf M., Doerr H.G., Hauffa B.P. et al.; Prevalence of autoantibodies associated with thyroid and celiac disease in Ullrich-Turner syndrome in relation to adult height after growth hormone treatment; J. Pediatr. Endocrinol. Metab. 2006:19, 149-154
81. Lleo A., Moroni L., Caliari L. et al.; Autoimmunity and Turner’s syndrome; Autoimmun. Rev. 2012:11, A538-543
82. Ostberg J.E., Attar M.J., Mohamed-Ali V. et al.; Adipokine dysregulation in turner syndrome: comparison of circulating interleukin-6 and leptin concentrations with measures of adiposity and C-reactive protein; J. Clin. Endocrinol. Metab. 2005:90, 2948-2953








