Endokrynol. Ped. 2015.14.1.50:67-70
DOI: 10.18544/EP-01.14.01.1513
Zespół Kallmanna – późne rozpoznanie i leczenie
Klinika Pediatrii, Endokrynologii, Diabetologii, Chorób Metabolicznych i Kardiologii Wieku Rozwojowego Pomorskiego Uniwersytetu Medycznego w Szczecinie
Słowa kluczowe: zespół Kallmanna, hipogonadyzm hipogonadotropowy
Streszczenie
Zespół Kallmanna jest jedną z przyczyn opóźnionego dojrzewania płciowego. Na obraz zespołu składają się hipogonadyzm hipogonadotropowy oraz upośledzenie lub brak węchu. Opisuje się także liczne towarzyszące cechy fenotypowe. Rozpoznanie najczęściej ustalane jest w okresie pokwitania, a nierzadko po 18 roku życia. Potwierdzenie genetyczne zespołu uzyskuje się zaledwie w 30% przypadków.
Wstęp
Opóźnione dojrzewanie płciowe u chłopców definiowane jest jako brak cech pokwitania po 14 rż. Zespół Kallmanna jest jedną z przyczyn opóźnionego dojrzewania płciowego. Częstość występowania szacowana jest na 1:10.000 [1]. Opisywane jest zarówno występowanie rodzinne, jak i sporadyczne. Klasyczny zespól Kallmana (HH1) jest dziedziczony w sposób autosomalnie dominujący, sprzężony z X – defekt w regionie Xp22.3 genu KAL 1 [1]. Potwierdzono także dziedziczenie autosomalne recesywne oraz zidentyfikowano mutacje w genach korelowanych z tym zespołem (np. FGFR1, PROK2, PROKR2, GNRHR) [2–4]. Genetyczne uwarunkowanie udaje się potwierdzić zaledwie w 20–30% przypadków [2,4]. Podstawowymi składowymi zespołu Kallmanna są hipogonadyzm hipogonadotropowy wynikający z izolowanego niedoboru GnRH oraz zaburzenia węchu (brak lub upośledzenie) [1,2]. Niedobór GnRH prowadzi do niskiej sekrecji gonadotropin (LH i FSH), co skutkuje nieprawidłowym wydzielaniem hormonów płciowych przez gonady. Pierwszym objawem tego zespołu, stwierdzanym już w okresie wczesnodziecięcym, jest małe prącie i wnętrostwo. Do innych objawów zespołu należą niedosłuch wady nerek rozszczep wargi i/lub podniebienia, wady linii pośrodkowej twarzy szczelina tęczówki, zaburzenia widzenia wady zgryzu, gotyckie podniebienie lustrzane ruchy rąk, niemożność płynnego śledzenia oczami, ataxia gynoidalna sylwetka, długie kończyny, krótszy tułów, wysoki wzrost owłosienie płciowe normalne lub osłabione ginekomastia wrodzona wada serca anomalie w układzie kostnym [2–7].
Opis przypadku
Chłopiec 17,5-letni został przyjęty do Kliniki z powodu opóźnionego dojrzewania płciowego. Pochodzi z ciąży IV, porodu III, urodzony o czasie, z masą ciała 2650 g, długością 57 cm, Apgar 10. Rozwój psychoruchowy w 1rż prawidłowy. W 4 rż uraz głowy bez utraty przytomności, złamanie kończyny górnej prawej. W 6 rż stwierdzono brak jąder w mosznie, wykonano orchidopeksję lewostronną, stwierdzono hypoplazję lewego jądra. Przeprowadzono także operację przepukliny pachwinowej po prawej stronie. W tym okresie u dziecka rozpoznano ADHD. Chłopiec został objęty opieką psychologiczno-logopedyczną. W kolejnych latach był kontrolowany przez lekarza rodzinnego. Od 16 rż pojawiły się drobne zmiany trądzikowe na nosie oraz owłosienie pachowe i łonowe bez innych objawów pokwitania. Poza skargami na opóźnione pokwitanie chłopiec zgłaszał obniżoną samoocenę z powodu różnic w budowie ciała w stosunku do rówieśników. Do tej pory poważnie nie chorował. Wywiad rodzinny obciążony: stan po operacji wędrujących jąder u ojca chłopca, choroby układu krążenia u dziadków.
Przy przyjęciu stan dobry, kontakt logiczny, wysoki tembr głosu. Wysokość ciała 3–10 centyl, masa ciała <3 centyla. Zaburzone proporcje ciała (długie kończyny, krótszy tułów). Sylwetka gynoidalna. Na skórze (obustronnie) pooperacyjne blizny w okolicy pachwinowej. Tkanka podskórna miernie rozwinięta – większa depozycja w obrębie brzucha i bioder; zaznaczona steatomastia. Gałki oczne osadzone prawidłowo. Źrenice równe, okrągłe, średniej wielkości, z prawidłową reakcją na światło. Podniebienie gotyckie. Klatka piersiowa asymetryczna. Narządy płciowe typu męskiego: mikropenis (2,5 cm), hipoplastyczna moszna, przedpokwitaniowe jądra (lewe 1 ml, prawe 1,5 ml), jądro prawe sprowadzalne z kanału pachwinowego. Skąpe owłosienie łonowe i pachowe (Ax II Pb II). Napięcie mięśniowe prawidłowe, siła mięśniowa prawidłowa, symetryczna. Układ kostny prawidłowy, zwiększony odstęp pomiędzy palcami stóp. Badanie nerwów czaszkowych: chłopiec nie czuje zapachów – dopytywany podaje, że „od zawsze” (poza aromatem mięty), zauważalny dyskretny oczopląs poziomy przy położeniach skrajnych oraz skokowe podążanie wzrokiem za przedmiotem w kierunku góra-dół. Poza tym w badaniu neurologicznym bez zmian.
W diagnostyce różnicowej brano pod uwagę konstytucjonalnie opóźnione dojrzewanie oraz hipogonadyzm hipo- i hipergonadotropowy, zespoły genetyczne przebiegające z zaburzeniami pokwitania, choroby przewlekłe. W wykonanych podstawowych badaniach dodatkowych bez odchyleń. Kliniczna i laboratoryjna eutyreoza. Prawidłowa zmienność dobowego rytmu kortyzolu i ACTH. Brak danych dla rozpoznania hiperprolaktynemii. W teście z LH-RH hipogonadyzm hipogonadotropowy (tab. I).
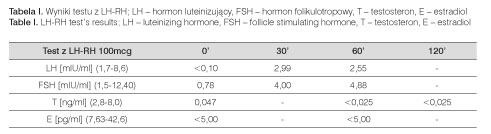
Wiek kostny kośćca przedramienia i ręki lewej jak dla chłopca w wieku 14 lat. Zapis EKG prawidłowy dla wieku. Usg jamy brzusznej bez odchyleń. W usg sutków nie stwierdzono obecności tkanki gruczołowej, w usg jąder echostruktura jednorodna. W wykonanym rezonansie magnetycznym mózgowia bez zmian. Przysadka mózgowa niepowiększona, sygnał części gruczołowej i nerwowej prawidłowy; wzmocnienie kontrastowe części gruczołowej jednorodne.
Przeprowadzono badania genetyczne. Uzyskano dodatni wynik na obecność sekwencji genu SRY i pozostałych genów w obrębie chromosomu Y (ZFY, AZFa, AZFb, AZFc); kariotyp prawidłowy: 46XY. Analiza techniką FISH – sonda Kallmann (Kal1) and Steroid Sulphatase Deficiency (STS) Probe Combination (Cytocell) – wykluczyła delecję w genie KAL1. Analiza sekwencji całego obszaru kodującego (wraz z fragmentami intronów sąsiadujących z eksonami) genu KAL1 (klasyczne sekwencjonowanie DNA metodą Sangera) nie potwierdziła zespołu Kallmana typu 1( HH1). W planie ewentualna analiza mutacji w innych genach korelowanych z tym zespołem (np. FGFR1, PROK2, PROKR2, GNRHR). Na podstawie całości obrazu klinicznego rozpoznano zespół Kallmanna. Do leczenia włączono preparat testosteronu w postaci iniekcji domięśniowych początkowo w dawce 50 mg co 4 tygodnie, stopniowo zwiększając dawkę. Obecnie w leczeniu 150 mg co 4 tygodnie. Po pięciu miesiącach leczenia zaobserwowano zmianę barwy głosu, zwiększenie wymiarów prącia (ob. 4 cm).
Pacjent został objęty poradnictwem genetycznym oraz przekazany do dalszej opieki endokrynologicznej.
Dyskusja
Zespół Kallmanna jest rzadkim schorzeniem. Częstość występowania szacowana jest zazwyczaj na 1:10.000 [1]. Zauważono, że występuje kilkukrotnie częściej u mężczyzn niż u kobiet. Cadman i wsp. podają, że choruje 1:8000–10000 mężczyzn i 1:40000–50000 kobiet [8]. Z uwagi na to, że zaburzenia genetyczne udaje się potwierdzić jedynie u ok. 30% chorych, rozpoznanie zespołu Kallmanna opiera się głównie na obrazie klinicznym [2].
Charakterystycznymi składowymi zespołu są hipogonadyzm hipogonadotropowy oraz zaburzenia węchu [1]. Często pierwsza manifestacja schorzenia to mikropenis i wnętrostwo, które rozpoznawane są w pierwszych latach życia [2]. U przedstawianego pacjenta wnętrostwo zostało stwierdzone w pierwszych latach życia i wymagało postępowania chirurgicznego w 6 rż. W kolejnych latach chłopiec był kontrolowany przez lekarza podstawowej opieki zdrowotnej. Pacjent podaje, że od około 14 roku życia niepokoił się brakiem postępu pokwitania, co zgłaszał lekarzowi rodzinnemu, który rozpoznał konstytucjonalnie opóźnione dojrzewanie. Zwraca uwagę fakt, że opisywany przez nas chłopiec miał wiele innych nieprawidłowości w badaniu przedmiotowym: zaburzone proporcje ciała (długie kończyny, krótszy tułów), gynoidalną sylwetkę, gotyckie podniebienie, asymetryczną klatkę piersiową, dyskretny oczopląs poziomy przy położeniach skrajnych oraz skokowe podążanie wzrokiem za przedmiotem w kierunku góra-dół. Podobne cechy opisywane są jako towarzyszące zespołowi Kallmanna [2–7]. Chory nie został jednak skierowany do specjalistycznej diagnostyki aż do ukończenia 17,5 lat.
Zaznaczyć należy, że opisywany pacjent jakość życia określił jako gorszą, głównie z powodu postrzegania go jako młodszego niż w rzeczywistości oraz przez pryzmat odnoszenia własnej („gorszej”) budowy ciała do rówieśników. Obniżona jakość życia, gorsza samoocena oraz negatywny wpływ na funkcjonowanie psychoseksualne także podnoszone jest co do tego zespołu w literaturze [9].
Warto podkreślić, że najczęściej podejrzenie zespołu Kallmanna wysuwane jest późno [2]. Zazwyczaj dopiero u starszych nastolatków z powodu opóźnionego dojrzewania płciowego – podobnie jak u naszego chorego. Czasem diagnostykę rozpoczyna się dopiero u młodych dorosłych. Postawienie rozpoznania na wcześniejszych etapach rozwoju wydaje się trudne. Mikropenis oraz wnętrostwo, często towarzyszące temu zespołowi, stwierdzane są jednak już w okresie wczesnodziecięcym. Należałoby zatem rozważyć wczesne objęcie tych dzieci opieką endokrynologiczną. Zespołowi mogą towarzyszyć liczne dodatkowe wady, m.in.: niedosłuch, wady serca, układu kostnego czy nerek [2]. Wczesna opieka specjalistyczna umożliwiłaby przeprowadzenie badań w tym kierunku, a także wcześniejsze ustalenie rozpoznania i włączenie leczenia substytucyjnego.
Postępowanie terapeutyczne u pacjentów z hipogonadyzmem hipogonadotropowym zależne jest od wieku oraz oczekiwań co do uzyskania płodności. Zazwyczaj leczenie rozpoczyna się od substytucji testosteronem [5]. Przedstawiany pacjent został poddany tej formie terapii. Po kilku miesiącach można podjąć próbę czasowego odstawienia testosteronu celem obserwacji, czy zostanie podjęte przez podwgórze spontaniczne wydzielanie GnRH [5]. Celem uzyskania płodności (indukcja spermatogenezy) najczęściej stosowane jest okresowe leczenie ludzką gonadotropiną kosmówkową (hCH – odpowiednik LH) z gonadotropiną menopauzalną (HMG – odpowiednik FSH), ewentualnie ludzką rekombinowaną FSH [5,9,10]. Spermatogenezę udaje się uzyskać u większości chorych (28–80%) [4,9–11]. Nie stosuje się monoterapii z użyciem hCH z uwagi na zwiększone ryzyko uszkodzenia krążenia wewnątrzjądrowego, zmian zapalnych i apoptozy komórek spermatogenezy, a nawet martwicy jąder [5]. Pulsacyjne, podskórne podawanie GnRH przy użyciu osobistej pompy także znajduje zastosowanie w skutecznym leczeniu zaburzeń spermatogenezy i pokwitania [4,5,9,10]. Z uwagi na koszty terapia GnRH wydaje się w obecnej sytuacji mało prawdopodobna. Po uzyskaniu potomstwa kontynuuje się leczenie testosteronem [5].
Warto podkreślić, że wczesne rozpoznanie zespołu pozwoliłoby na wdrożenie postępowania terapeutycznego we wcześniejszym wieku. Umożliwiłoby to prawidłowy rozwój oraz być może poprawiło samoocenę i jakość życia osób z zespołem Kallmanna.
Piśmiennictwo
1. Laitinen E.M., Vaaralahti K., Tommiska J. et al.; Incidence, phenotypic features and molecular genetics of Kallmann Syndrome in Finland; Orphanet J. Rare Dis. 2011:6, 41, [dostęp 14.02.2014] http://www.ojrd.com/content/6/1
2. Kaplan J.D., Bernstein J.A., Kwan A. et al.; Clues to an early diagnosis of Kallmann syndrome; Am. J. Med. Genet. Part A 2010:152A, 2796-2801
3. Cadman S.M., Kim S.H., Hu Y. et al.; Molecular pathogenesis of Kallmann’s syndrome; Horm. Res. 2007:67, 231-242
4. Kaur K.K., Allahbadia G.N., Singh M.; Male hypogonadism-areview of secondary hypogonadism with special emphasis on hypogonadotropic hypogonadism; J. Endocrinol. Diabetes Obes. 2014:2(2), 1023
5. Rabijewski M., Zgliczyński W.; Etiopatogeneza, rozpoznawanie i leczenie hipogonadyzmu u mężczyzn; Pol. J. Endocrinol. 2009:60, 222-233
6. John H., Schmid C.; Kallmann’s syndrome: clues to clinical diagnosis; Int. J. Impot. Res. 2000:12, 269-271
7. Klatka M., Szewczyk L.; Zespół Kallmanna. Prezentacja przypadku 17-letniej pacjentki; Pediatr. Endocrinol. 2009:8, 3(28), 61-64
8. Sowińska-Przepiera E., Andrysiak-Mamos E., Syrenicz A.; Hipogonadyzm. Endokrynologia w codziennej praktyce lekarskiej; Red. Syrenicz A. Wydaw. Pomorskiej Akademii Medycznej w Szczecinie 2009, 391-405
9. Dwyer A.A., Quinton R, Pitteloud N et al.; Psychosexual Development in Men with Congenital Hypogonadotropic Hypogonadism on Long-Term Treatment: A Mixed Methods Study; Sex. Med. 2014 [dostęp 16.02.2014] http://onlinelibrary.wiley.com/doi/10.
10. Farshchi H., Shahnazi A., Azizi F.; Effects of Testosterone and Gonadotropin Therapy in Men with Hypogonadotropic Hypogonadism; Int. J. Endocrinol. Metab. 2009:4, 242-247
11. Sato N., Hasegawa T., Hori N. et al.; Gonadotrophin therapy in Kallmann syndrome caused by heterozygous mutations of the gene for fibroblast growth factor receptor 1: report of three families: case report; Hum. Reprod. 2005:20(8), 2173-2178








