Endokrynol. Ped. 13/2014;1(46):31-40
DOI: 10.18544/EP-01.13.01.1475
Rola limfocytów T γδ w patogenezie cukrzycy typu 1
Katedra i Klinika Endokrynologii i Diabetologii Wieku Rozwojowego, Uniwersytet Medyczny we Wrocławiu
Słowa kluczowe: limfocyty Tγδ, cukrzyca typu 1, patogeneza
Streszczenie
Cukrzyca typu 1 (DM1) wynika ze zniszczenia komórek β wysp trzustkowych przez limfocyty T w czasie przewlekłego procesu autoimmunologicznego. Wystąpienie autoagresji jest związane z predyspozycją genetyczną oraz działaniem inicjujących czynników środowiskowych. Limfocyty T γδ uczestniczą w mechanizmach obrony przeciwnowotworowej oraz przeciwzakaźnej, uważa się, że mogą brać udział w zapobieganiu rozwoju insulitis. Wykazano korelację między liczbą limfocytów T γδ a stopniem upośledzenia pierwszej fazy wydzielania insuliny w grupie prediabetes, co wydaje się potwierdzać regulatorową rolę tych limfocytów w rozwoju DM1
Wstęp
Mimo że historia badań nad rolą limfocytów Tγδ w cukrzycy typu 1 rozpoczęła się w połowie lat dziewięćdziesiątych ubiegłego wieku, to ich znaczenie w patogenezie chorób autoimmunologicznych, w tym cukrzycy typu 1, pozostaje nadal słabo poznane, a wyniki przedstawianych badań są niejednolite. Populacja limfocytów Tγδ w odpowiedzi immunologicznej może działać zarówno pro-, jak i przeciwzapalnie. Wiąże się to z różnicami w budowie łańcucha gamma i delta receptora TCR, determinującymi funkcję komórki: cytotoksyczną lub regulatorową, ale również powinowactwem omawianych limfocytów do różnych tkanek [1,2]. Opisywane są zaburzenia funkcji limfocytów Tγδ i wielkości ich populacji we krwi obwodowej oraz nacieczenie narządów przez te komórki w rozmaitych chorobach z autoagresji, m.in. w stwardnieniu rozsianym, toczniu układowym, jak również w celiakii [3–7].
Zaburzenie mechanizmów immunologicznych w rozwoju cukrzycy typu 1
Klasyczna teoria odporności zakłada, że układ immunologiczny zabezpiecza organizm przed wszystkimi obcymi antygenami, a podstawę odpowiedzi autoimmunizacyjnej stanowi utracona zdolność układu odpornościowego do odróżnienia antygenów „własnych” od „obcych”. Z kolei według modelu sygnału zagrożenia Polly Matzinger inicjacja swoistej odpowiedzi immunologicznej zależy od rozpoznania nie tylko obcej natury antygenu, ale także warunków, w jakich dany antygen jest prezentowany komórkom immunokompetentnym. Jeśli dane warunki zostaną odebrane przez organizm jako patologiczne, sprzyjać będą powstaniu odpowiedzi immunologicznej bez względu na to, czy antygen jest własny, czy obcy. Komórki prezentujące antygen (APC, antigen presenting cells) muszą więc otrzymać dodatkowy „sygnał zagrożenia”, aby rozpocząć odpowiedź immunologiczną. W sytuacji autoagresji takim sygnałem może być nieswoisty proces zapalny towarzyszący uwalnianiu autoantygenów w wyniku zniszczenia tkanek [8]. Teoria Matzinger wpisuje się w model patogenezy cukrzycy typu 1, stworzony przez Nerupa i wsp. Zakłada on, że po uszkodzeniu wysp trzustkowych przez czynniki inicjujące autoantygeny są prezentowane przez komórki APC limfocytom T pomocniczym CD4+ (Th, T helper), co prowadzi do ich aktywacji. Aktywne limfocyty Th za pośrednictwem produkowanych przez siebie limfokin indukują apoptozę/nekrozę komórek beta trzustki, powodując naciek wysp trzustkowych komórkami mononuklearnymi (insulitis) [9]. Według twórców tej teorii w procesie indukowania autoantygenów na powierzchni komórek β biorą udział czynniki wewnętrzne (cytokiny, wolne rodniki) lub zewnętrzne (toksyny, wirusy), jednak proces autoimmunologiczny jest inicjowany w komórkach beta wysp Langerhansa.
Podkreśla się rolę defektu mechanizmów regulacyjnych, w tym nieprawidłowej selekcji autoantygenów w grasicy w powstawaniu autoagresji.
Jednym z istotnych mechanizmów regulacji odpowiedzi układu immunologicznego są limfocyty T-regulatorowe (Treg). Zróżnicowana fenotypowo populacja limfocytów Treg bierze udział m.in. w odpowiedzi przeciwinfekcyjnej, przeciwnowotworowej i poszczepiennej, wytwarzaniu tolerancji na antygeny pokarmowe, bakterie saprofityczne i transplantacyjne, hamuje limfocyty autoreaktywne. Przypuszcza się, że wystąpienie cukrzycy może mieć związek z redukcją liczby limfocytów Treg oraz osłabieniem ich aktywności supresyjnej i/lub jest wynikiem pojawienia się opornych na regulację patogennych limfocytów T [10]. Według Piccirillo nieprawidłowe działanie m.in. limfocytów regulatorowych może mieć kluczowe znaczenie w inicjowaniu zaburzonych procesów immunologicznych, w wyniku których dochodzi do załamania się autotolerancji i postępującego zniszczenia komórek β wysp trzustkowych [11,12].
Badania wykazały, że wydzielana przez komórki β insulina może być autoantygenem inicjującym kaskadę immunologiczną z udziałem m.in. limfocytów T, w konsekwencji której dochodzi do rozwoju cukrzycy typu 1 [13]. Podanie myszom NOD insuliny syntetycznej, doustnie, donosowo czy w postaci aerozolu, hamowało lub opóźniało początek reakcji immunologicznej i zmniejszało częstość zachorowań na cukrzycę u badanych zwierząt. Dośluzówkowe podanie insuliny jako pojedynczego autoantygenu może bowiem indukować tolerancję immunologiczną przeciwko patogennym komórkom T swoistym wobec kilku autoantygenów. Fenomen ten jest prawdopodobnie wynikiem aktywacji komórek regulatorowych, do których zalicza się limfocyty T γδ [13].
Limfocyty T γδ
Limfocyty T mają na swojej powierzchni receptor transmembranowy TCR (T Cell Receptor), biorący udział w kluczowym momencie inicjacji odpowiedzi autoimmunologicznej, jakim jest rozpoznanie antygenu. Ze względu na budowę wyróżnia się dwie populacje limfocytów T: limfocyty T αβ pomocnicze i supresorowe/cytotoksyczne oraz limfocyty T γδ pomocnicze i supresorowe/cytotoksyczne [14].
W połowie lat osiemdziesiątych dwudziestego wieku Tonegawa z zespołem opublikowali wyniki swoich analiz molekularnych, prezentując gen kodujący łańcuch γ receptora TCR [14]. W ślad za tym odkryciem ujawniono istnienie receptora TCR γδ i wykryto, że gen dla łańcucha δ tego receptora znajduje się wewnątrz locus genu dla łańcucha α receptora TCR αβ [15].
Limfocyty T γδ są stosunkowo liczne w skórze, płucach i nabłonku jelitowym, co wskazuje na ich udział w mechanizmach obronnych w miejscach kontaktu z obcym antygenem. Stanowią one pierwszą linię obrony w zakażeniach wywołanych przez rodzinę Mycobacteriacae. W płucach wykazują swoistość wobec Mycobacterium tuberculosis, są zdolne do proliferacji w obecności białek szoku termicznego (heat shock proteins, HSPs), głównie HSP-60 i HSP-65, których ekspresja jest odpowiedzią na zakażenie prątkiem gruźlicy. Makrofagi płucne kostymulują tę proliferację, prezentując komórkom T γδ żywe mykobakterie. Dodatkowo limfocyty T γδ są pobudzane przez inny ligand mykobakteryjny: białko o wielkości 2-10 kDa, oporne na działanie enzymów proteolitycznych i wiążące się z lektynami [16].
Opisuje się zdolność tych komórek do rozpoznawania białek szoku termicznego i innych niskomasowych antygenów, a także autoantygenów. Posiadają one właściwości cytotoksyczne i cechy charakterystyczne dla komórek NK, „naturalnych zabójców” [17].
Większość limfocytów T γδ stanowią komórki Vγ9/Vδ2, odpowiedzialne za obroną przeciwzakaźną i posiadające niezwykłą zdolność do uaktywniania odpowiedzi zarówno Th1, jak i Th2 zależnej. Według Caccamo i wsp. zmniejszenie liczby tej populacji może zwiększać ryzyko choroby autoimmunologicznej po trzydziestym roku życia [18].
Limfocyty T γδ stanowią w organizmie człowieka około 4% puli limfocytów T, ich ilość jest prawdopodobnie stała w czasie życia człowieka [19]. Budowa łańcucha gamma i delta receptora TCR determinuje powinowactwo do różnych tkanek (skóra Vγ3/Vδ1, język i pochwa Vγ4/Vδ1, płuca Vγ2/Vδ5, jelita Vγ5 z Vδ4, Vδ5, Vδ6 i Vδ7, krew i limfa Vγ2 z Vδ5, Vδ6 i Vδ7) oraz funkcję tych komórek, które uczestniczą w reakcjach obronnych przed infekcjami wirusowymi, bakteryjnymi i pasożytniczymi (Vγ9/Vδ2) i cytolizie autologicznych komórek rakowych (VγX/Vδ1). Selektywne wytwarzanie limfocytów T γδ jest prawdopodobnie niezbędne w powstawaniu efektywnych barier immunologicznych [20].
W przeciwieństwie do limfocytów T αβ, limfocyty T γδ nie wymagają obecności antygenów klasy II HLA w procesie rozpoznawania antygenu, natomiast są zdolne do rozpoznania antygenów przez receptor TCR w kooperacji z klasą antygenów Ib, obejmującą cząsteczki CD1, Qa oraz H2-M3. Wykazują ekspresję antygenów powierzchniowych CD 16 oraz CD 56 charakterystycznych dla komórek NK. Dzięki temu mogą uczestniczyć w zjawisku spontanicznej cytotoksyczności komórkowej zależnej od przeciwciał. Uważa się, że jest to populacja limfocytów o cechach pośrednich między limfocytami T i komórkami NK, stanowiąca pomost między odpornością swoistą i nieswoistą [21].
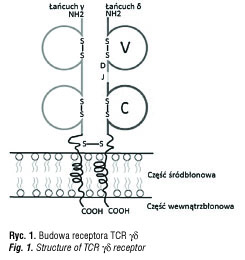
Budowa receptora TCR
Receptor TCR jest heterodimerem o masie 90 kDa. Jest zbudowany z dwóch łańcuchów polipeptydowych alfa i beta (TCR αβ, TCR 2) lub gamma i delta (TCR γδ, TCR 1) [21,22]. W błonie komórkowej limfocytów łańcuchy polipeptydowe alfa/beta lub gamma/delta połączone są z kompleksem CD3, niezbędnym do wewnątrzkomórkowego przekazywania sygnału i prawidłowego rozpoznawania antygenów. Cząsteczka CD3 zbudowana jest z podjednostek γ, δ, ε, ζ i η. Budowę receptora TCR γδ przedstawia rycina 1.
Rozwój limfocytów T γδ
Wytworzenie takich limfocytów T, które będą miały prawidłowo zbudowany receptor TCR i nie będą rozpoznawały własnych antygenów, jest głównym celem procesów zachodzących w grasicy. Prekursory limfocytów T napływają do grasicy już w 7–8 tygodniu ciąży. Istnieją co najmniej dwa rodzaje komórek zasiedlających grasicę. Jedne z nich są bezpośrednimi prekursorami tymocytów (komórki pre-T), drugi typ komórek, określany jako wspólne komórki progenitorowe limfopoezy (common lymphocyte progenitors, CLP), jest zdolny do różnicowania nie tylko w limfocyty T, ale także w limfocyty B, komórki NK oraz komórki dendrytyczne [22].
W trakcie dojrzewania tymocytów następuje kilka procesów selekcji, które można podzielić na dwie fazy: 1. Faza wczesna, w której dojrzewające komórki nie posiadają receptorów rozpoznających antygen. Rozpoczyna się ona od powstania komórek progenitorowych w szpiku i obejmuje zasiedlenie grasicy, ekspansję komórek zasiedlających, ukierunkowanie rozwoju w stronę wczesnych tymocytów, rearanżację genów dla TCR, selekcję β oraz wyłączenie alleliczne.
2. Faza późna, w której tymocyty posiadające pełną ekspresję receptorów rozpoznających antygen poddawane są selekcji pozytywnej i negatywnej.
Najmniej dojrzałe tymocyty są określane jako komórki potrójnie negatywne (TdT+/- CD3-CD4-CD8-CD34+CD44+CD117+HLA-DR+/-), ponieważ nie posiadają one ani kompleksu TCR/CD3, ani cząsteczek CD4 czy CD8. W kolejnych etapach dojrzewania tymocytów na ich błonach pojawia się różny układ antygenów, co pozwala wyróżnić subpopulacje protymocytów odpowiadające kolejnym etapom dojrzewania tych komórek. W wyniku kontaktu komórek TdT+CD1+/-CD25+ ze zrębem grasicy dochodzi do intensywnej proliferacji. Do rozpoczęcia procesu rearanżacji genów kodujących receptor TCR niezbędna jest jak największa pula komórek, ponieważ im więcej komórek przystąpi do tego procesu, tym większa będzie różnorodność receptorów. W każdej komórce rearanżacja odbywa się niezależnie i doprowadza do wytworzenia przypadkowych kombinacji genów VDJ (odpowiedzialnych za łańcuch delta i beta) oraz VJ (łańcuch gamma i alfa). Receptory TCR γδ powstają w filogenezie wcześniej niż receptory TCR αβ. Dodatkowo do ich syntezy wykorzystywane są niektóre z genów VJ, co ogranicza różnorodność receptorów TCR. Czynnikiem przyspieszającym proliferację protymocytów w kierunku tymocytów jest czynnik wzrostu komórek macierzystych SCF (stem cel factor, ligand Kit), działający synergistycznie z interleukiną 7 i interleukiną 12 [22].
Na powierzchni wczesnych tymocytów pojawiają się cząsteczki CD1 i CD3, zanikają zaś CD25 i CD117. Tzw. późne tymocyty (CD1+/-CD2+CD3+CD5+CD7+) podlegają w grasicy selekcji pozytywnej i negatywnej, w wyniku której wyłoniona zostaje pula limfocytów T prawidłowo rozpoznających obce antygeny. Proces selekcji pozytywnej ma na celu wyłonienie tych komórek, które prawidłowo rearanżują geny kodujące łańcuch beta (selekcja β) i cały receptor TCR (po rekombinacji genów dla łańcucha alfa). Na tym etapie dojrzewania tymocytów αβ dochodzi do tzw. restrykcji MHC, czyli uwarunkowania rozpoznawania przez limfocyty T antygenów prezentowanych przez własne cząsteczki MHC. Limfocyty, które będą rozpoznawać antygeny przez MHC klasy I, zachowują ekspresję CD8, natomiast wiążące antygen przez MHC klasy II pozostaną CD4+. Selekcja negatywna wyklucza tymocyty, które ze zbyt dużym powinowactwem rozpoznają własne antygeny. Dodatkowo tymocyty γδ „uczą się” rozpoznawać własne białka organizmu. Część z nich podlega także selekcji poza grasicą, prawdopodobnie w jelicie [22].
W selekcji tymocytów biorą udział komórki nabłonkowe kory i rdzenia grasicy. Są one zdolne do prezentacji wielu narządowo swoistych antygenów, np. insuliny. Za ekspresję tych białek jest odpowiedzialny czynnik transkrypcyjny AIRE (autoimmune regulator). Brak prawidłowego czynnika AIRE jest przyczyną autoimmunologicznego zespołu niedoczynności wielogruczołowej (Autoimmune Polyglandular Syndrome, APS), w którym brak ekspresji antygenów narządowo swoistych w grasicy jest przyczyną nieskutecznej selekcji negatywnej [23].
Należy podkreślić, że w syntezie i rearanżacji genów dla receptora TCR istnieje przypadkowość, możliwe jest zatem powstanie receptorów TCR niespełniających żadnej pozytywnej roli, a nawet zdolnych do autoagresji. Tymocyty γδ różnicują się w kierunku trzech subpopulacji dojrzałych limfocytów T γδ, zasilających pulę limfocytów T na obwodzie. Subpopulacje limfocytów T γδ dzieli się ze względu na antygeny powierzchniowe na: 1) limfocyty podwójnie negatywne, niewykazujące na swojej powierzchni ekspresji antygenów CD4 i CD8 (limfocyty T γδ CD4-CD8-); 2) posiadające jedynie antygen CD8 (limfocyty T γδ CD4-CD8+) oraz 3) posiadające tylko antygen CD4 (limfocyty T γδ CD4+CD8-) [22].
Czynniki wpływające na rozwój limfocytów T γδ
Homeostaza limfocytów T γδ jest zależna od limfocytów T αβ, komórek dendrytycznych i NK, które oddziaływają na omawianą populację komórek T przez interleukinę 7 (IL-7) oraz interleukinę 15 (IL-15). Największą rolę w regulacji wielkości populacji komórek T γδ i możliwości ich ekspansji odgrywają limfocyty T αβ, prawdopodobnie ze względu na wielkość tej populacji [19].
Limfocyty T γδ są zdolne do wydzielania cytokin, m.in.: interleukiny 2 (IL-2), interleukiny 10 (IL-10), interleukiny 12 (IL-12), interleukiny 15 (IL-15), interleukiny 17 (IL-17) i interferonu γ (IFN-γ) [24].
Aktywność przeciwnowotworowa limfocytów T γδ
Wykazano, że limfocyty T γδ uczestniczą w procesie obrony przeciwnowotworowej i przeciwzakaźnej na drodze perforynozależnej cytolizy: uwolnione z ziarnistości granzymy i perforyny prowadzą do zniszczenia zainfekowanych komórek gospodarza. Inny mechanizm przeciwnowotworowego i przeciwzakaźnego działania komórek Tγδ polega na wydzielaniu cytokin – mediatorów reakcji zapalnej. Do najważniejszych należy granulocytarno-makrofagowy czynnik wzrostu (granulocyte-macrophage colony-stimulating factor, GM-CSF), który zwiększa migrację neutrofili do miejsca infekcji, i transformujący czynnik wzrostu beta (transforming growth factor β, TGF-β), biorący udział w aktywacji limfocytów B. Limfocyty T γδ wydzielają interleukinę 2 (IL-2) wzmagającą aktywność limfocytów T, interferon γ (IFN-γ), pobudzający makrofagi i przyspieszający proces prezentacji antygenu, oraz interleukinę 10 (IL-10) przyspieszającą resorpcję zmian zapalnych [25]. Nowotwory powodują aktywację tych komórek poprzez ekspresję mitochondrialnej F1-ATPazy, która w połączeniu z apolipoproteiną A-I jest rozpoznawana przez receptor komórki T Vγ9/Vδ2. Komórki T γδ wykazują się wysoką aktywnością przeciwnowotworową w chłoniakach nieziarniczych oraz w szpiczaku mnogim. Eliminują one także komórki charakteryzujące się wysoką ekspresją białka szoku termicznego HSP-70 lub jego homologów [26].
Do eliminacji komórek nowotworowych dochodzi głównie poprzez pobudzenie TCR z udziałem antygenu Qa-Tla bądź na drodze innych mechanizmów nieangażujących TCR. Komórki T γδ CD3+CD16+ mogą uczestniczyć w zjawisku spontanicznej cytotoksyczności komórkowej zależnej od przeciwciał (antibody-dependent cellular cytotoxicity, ADCC). Rozpoznawanie komórek nowotworowych przez limfocyty T γδ odbywa się niezależnie od antygenów MHC. Zasada ta prawdopodobnie nie dotyczy grupy limfocytów naciekających nowotwór (tumor infiltrating lymphocytes, TIL).
W przypadku guza Wilmsa, raka nerki, sutka, jajnika, trzustki, wątroby, jelita grubego, w czerniaku złośliwym i mięsakach obserwowana cytotoksyczność jest uzależniona od białek MHC [27]. Limfocyty T γδ CD69+ charakteryzują się wzmożoną ekspresją czynnika martwicy nowotworów α (tumor necrosis factor α), co przekłada się na ich funkcję ochronną przeciw infekcjom, chorobom autoimmunologicznym i nowotworom [28]. Interesujące wydają się spostrzeżenia Sowińskiej i wsp., dotyczące roli subpopulacji limfocytów T γδ u chorych na chłoniaki nieziarnicze i szpiczaka mnogiego. Według autorów większy odsetek limfocytów T γδ oraz aktywowanych limfocytów T γδ CD25+ i T γδ CD69+ występujący u chorych przed leczeniem w porównaniu ze zdrowymi zdaje się świadczyć o aktywacji mechanizmów obrony przeciwnowotworowej i może być uznany za czynnik dobrego rokowania [29].
Rola limfocytów T γδ w cukrzycy typu 1
Na zwierzęcym modelu cukrzycy typu 1 dowiedziono, iż limfocyty T γδ mogą brać udział w zapobieganiu rozwoju insulitis. Według hipotezy Harrisona limfocyty Tγδ miałyby być mediatorami tolerancji immunologicznej wywodzącej się ze śluzówkowego układu immunologicznego i/lub – analogicznie do działania limfocytów wewnątrznabłonkowych (IELs, intraepithelial lymphocytes) na komórki śluzówki jelita – hamować apoptozę komórek β trzustki i promować ich regenerację [13,30]. Uznali oni, że niedobór tych komórek skutkuje ujawnieniem się cukrzycy typu 1, a ich przeszczepienie zapobiega rozwojowi autoagresji u biorcy [omówione w 13]. Obserwacje te zostały potwierdzone w modelu zwierzęcym: czynnikiem, który zabezpieczał myszy NOD przed rozwojem cukrzycy, była niewielka liczba aktywowanych limfocytów Tγδ CD8+ [31–34]. Limfocyty cytotoksyczne/supresorowe Tγδ CD8+ i „podwójnie negatywne” limfocyty Tγδ CD4-CD8- cechują się dużą swoistością antygenową i wysokim potencjałem supresyjnym. Wykazano, że 2,5x103 tych limfocytów może zahamować 7x107 komórek efektorowych. Limfocyty te charakteryzuje wysoka ekspresja antygenu Fas. Po połączeniu antygenu Fas na limfocycie Tγδ z ligandem FasL na limfocycie Tαβ dochodzi do przekazania sygnału wywołującego apoptozę komórki efektorowej. Jest to forma pozytywnej immunoregulacji, pozwalająca na utrzymanie homeostazy mechanizmów obronnych [35]. Wobec tego nie dziwi fakt, że podanie myszom NOD niewielkiej liczby komórek Tγδ CD8+ pochodzących od zwierząt wcześniej immunizowanych insuliną donosową lub doustną powoduje opóźnienie rozwoju insulitis lub całkowite zahamowanie tego procesu. Według Hoffmanna i Nanno budowa receptora TCR γδ pozwala bowiem na rezydowanie tych komórek w śluzówkach, np. w błonie śluzowej nosogardzieli i przewodu pokarmowego, co skutkuje „ułatwionym” dostępem do różnych antygenów (w cytowanych pracach – na podawaną donosowo lub doustnie insulinę), a więc zwiększeniem możliwości wytworzenia tolerancji na te antygeny. Na tej samej zasadzie limfocyty Tγδ zabezpieczały badane zwierzęta przed rozwojem innych niż cukrzyca chorób autoimmunologicznych [36–38]. Badania Goldratha i wsp. dowiodły, że u myszy NOD odsetek limfocytów Tγδ, szczególnie „podwójnie pozytywnych” Tγδ CD4+CD8+ i „podwójnie negatywnych” Tγδ CD4-CD8-, był większy wśród komórek naciekających węzły chłonne w porównaniu do ich odsetka we krwi [39].
Krętowski i wsp. porównywali populację limfocytów T γδ u pacjentów z cukrzycą typu 1 oraz u ich krewnych z obecnymi autoprzeciwciałami (grupa prediabetes) [40,41]. Grupa pacjentów z cukrzycą typu 1 liczyła 22 osoby w wieku 19,1 ± 6,8 lat. Chorobę rozpoznano u nich 3–6 miesięcy przed badaniem limfocytów Tγδ, była ona dobrze kontrolowana, nie odnotowano przypadków kwasicy ketonowej. Grupę prediabetes stanowiło 26 krewnych pierwszego stopnia pacjentów z cukrzycą typu 1, u których utrzymywało się wysokie miano przeciwciał ICA (> 20 jednostek JDF) bez typowych dla cukrzycy objawów i braku biochemicznych kryteriów cukrzycy wg WHO. U 20/23 pacjentów obecne były oprócz ICA inne przeciwciała przeciwwyspowe, najczęściej GADA (17/20 pacjentów). Grupa kontrolna składała się ze zdrowych ochotników bez zaburzeń gospodarki węglowodanowej i chorób autoimmunologicznych. Stwierdzono, że populacja limfocytów Tγδ u pacjentów z prediabetes i z cukrzycą typu 1 nie różniła się istotnie statystycznie, jednak odsetek badanych komórek u pacjentów z cukrzycą i w grupie prediabetes był mniejszy niż w grupie kontrolnej. Ponadto u krewnych chorych na cukrzycę z obecnymi przeciwciałami i upośledzonym wydzielaniem insuliny odsetek limfocytów Tγδ był istotnie mniejszy w porównaniu z grupą kontrolną [40].
Niejednoznaczne obserwacje przedstawili Lang i wsp., którzy porównywali populacje limfocytów Tγδ w następujących grupach badawczych: u 14 pacjentów ze świeżo rozpoznaną cukrzycą typu 1, u krewnych pierwszego i drugiego stopnia chorych na cukrzycę typu 1, w tym u 26 osób, u których nie wykryto przeciwciał ICA, oraz 23 pacjentów ICA - pozytywnych, u 9 pacjentów z cukrzycą trwającą średnio 7, 2 lata. Grupę kontrolną stanowili ochotnicy, u których nie stwierdzono obecności przeciwciał ICA, a ich wywiad rodzinny był nieobciążony żadną chorobą autoimmunologiczną. Zaobserwowano większy odsetek limfocytów Tγδ u krewnych chorych na cukrzycę z obecnymi w wysokim mianie przeciwciałami przeciwwyspowymi ICA. Wielkości populacji limfocytów Tγδ u pacjentów ze świeżo rozpoznaną cukrzycą typu 1, a także u tych krewnych, u których nie stwierdzono obecności autoprzeciwciał, nie różniły się od wielkości populacji badanych komórek u zdrowych. Zwiększenie populacji limfocytów Tγδ wynikało ze zwiększenia frakcji T Vγ9/Vδ2 [42,43]. Z drugiej strony autorzy stwierdzili, że w grupie badanych z wysokim ryzykiem rozwoju cukrzycy typu 1 (z obecnymi autoprzeciwciałami) u osób, u których sekrecja insuliny była już zaburzona, odsetek limfocytów Tγδ był mniejszy w porównaniu do osób z prawidłowym wydzielaniem tego hormonu [42]. Dalsza obserwacja tych pacjentów wykazała, że zmniejszanie się populacji limfocytów Tγδ korelowało z upośledzeniem wydzielania insuliny i ujawnieniem się cukrzycy typu 1, podczas gdy u osób, u których odsetek omawianych limfocytów nie zmniejszał się, funkcja komórek β pozostawała prawidłowa. Lang dowiódł ponadto, że komórki T Vγ9/Vδ2 biorą udział jako komórki regulatorowe w rozwoju cukrzycy typu 1 i monitorowanie ich odsetka może użytecznym dodatkowym czynnikiem prognostycznym jej rozwoju [42,43].
Zarówno Lang, jak i Krętowski wykazali, że pacjenci z prediabetes, u których populacja limfocytów Tγδ była największa, cechowali się prawidłową sekrecją insuliny [40,42].
Zespół Pujol-Borrel oceniając populację limfocytów Tγδ w różnych endokrynopatiach autoimmunologicznych, m.in. w cukrzycy typu 1 (23 dorosłych pacjentów z chorobą trwającą krócej niż 6 miesięcy), spośród których u 13/23 (56%) obecne były przeciwciała ICA), stwierdził, że odsetek komórek Tγδ we krwi obwodowej pacjentów z cukrzycą nie różnił się od odsetka tych limfocytów w grupie kontrolnej, nie zależał od wieku chorych ani obecności przeciwciał ICA w osoczu [44].
Z kolei w badaniach Gyarmati i wsp. populacja limfocytów Tγδ u chorych na cukrzycę typu 1 i u ICA –pozytywnych krewnych pacjentów z cukrzycą typu 1 (grupa prediabetes) była istotnie większa niż w grupie kontrolnej, a frakcje badanych komórek w grupie prediabetes i u pacjentów ze świeżo rozpoznaną cukrzycą typu 1 były porównywalne [45].
Odmienne rezultaty zacytowanych badań mogą być wynikiem różnego wieku analizowanych chorych, czasu jaki upłynął od rozpoznania do wykonania oznaczeń, jak również różnych stadiów zaawansowania insulitis u tych pacjentów. Doświadczenia przeprowadzone w różnych chorobach autoimmunologicznych wykazały bowiem akumulację limfocytów Tγδ w zajętych organach, np. w płynie maziówkowym i maziówce chorych na reumatoidalne zapalenie stawów, w okolicy okołonaczyniowej naczyń skóry u osób z twardziną systemową oraz w tkankach obwodowych w toczniu trzewnym [46–49]. Nieprawidłowa dystrybucja tej frakcji limfocytów mogła skutkować zmniejszeniem się odsetka komórek we krwi obwodowej. Potwierdzeniem tej koncepcji jest m.in. praca Robak i wsp., którzy przebadali 32 chorych na tocznia trzewnego i wykazali, że bezwzględna liczba limfocytów Tγδ we krwi obwodowej jest istotnie mniejsza niż u zdrowych dawców [5]. Interesujący jest fakt, że również w przypadku pacjentów z chorobą nowotworową odsetek limfocytów Tγδ we krwi obwodowej był mniejszy w porównaniu do osób zdrowych [50–52]. Zgodnie z teorią migracji limfocytów Tγδ w chorobach nowotworowych, zaproponowaną przez Zocciego, są one bowiem przyciągane przez antygeny nowotworowe, przemieszczają się z krwi obwodowej do tkanki, w której rozwija się nowotwór. Tam stanowią pierwszą linię obrony przeciwnowotworowej, dzięki swojej aktywności cytolitycznej eliminując komórki transformowane nowotworowo [50,53]. Wydaje się, że rezultaty badań w chorobach autoimmunologicznych mogą przemawiać za słusznością tej hipotezy również w chorobach z autoagresji.
Poznanie zaburzonych mechanizmów immunologicznych prowadzących do rozwoju insulitis i zniszczenia komórek naturalnie produkujących jeden z najważniejszych życiowych hormonów, w tym zbadanie swoistych funkcji limfocytów Tγδ w rozwoju cukrzycy, jest jednym z podstawowych zadań w aspekcie leczenia tej choroby.
Piśmiennictwo
1. Bhagat G., Naiyer A.J., Shah J.G., Harper J. et al.; Small intestinal CD8+TCR gamma delta+ NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease; J. Clin. Invest. 2008:118, 281-293
2. Kühl A.A., Pawlowski N.N., Grollich K., Blessenohl M. et al.; Human peripheral gamma delta T cells possess regulatory potential; Immunology 2009:128, 580-588
3. Ziegler H.K.; The role of gamma/delta T cells in immunity to infection and regulation of inflammation; Immunol. Res. 2004:29, 293-302
4. Kapp J.A., Kapp L.M., McKenna K.C.; Gammadelta T cells play an essential role in several forms of tolerance; Immunol. Res. 2004:29, 93-102
5. Robak E., Błoński J.Z., Bartkowiak J.,Niewiadomska H. et al.; Circulating TCR γδ cells in thepatients with systemic lupus erythematosus; Mediators of Inflammation 1999:8, 305-312
6. Dandekar A.A., Perlman S.; Virus-induced demyelination in nude mice is mediated by gamma delta T cells; Am. J. Pathol. 2002:161, 1255-1263
7. Xu Y., Kapp J.A.; γδ T cells are critical for the induction of anterior chamber-associated immune deviation; Immunology 2001:104, 142-148
8. Matzinger P.; Tolerance, danger and the extender family; Annu. Rev. Immunol. 1994:12, 991-1045
9. Nerup J., Mandrup-Poulsen T., Helqvist S., Andersen H.U. et al.; On the pathogenesis of IDDM; Diabetologia 1994:37 Suppl 2, S82-89
10. Gregori S., Giarratana N.M., Smiroldo S., Adorini L.; Dynamics of pathogenesic and suppressor T cells in autoimmune diabetes development; J. Immunol. 2003:171, 4040-4047
11. Piccirillo C.A., Thornton A.M.; Cornerstone of peripheral tolerance: naturally occurring CD4+CD25+ regulatory T cells; Trends. Immunol. 2004:25, 374-380
12. Li C.R., Baaten B.J., Bradley L.M.; Harnessing memory adaptive regulatory T cells to control autoimmunity in type 1 diabetes; J. Mol. Cell. Biol. 2012:4, 38-47
13. Hänninen A., Harrison L.C.; γδ T cels as mediators of mucosal tolerance: the autoimmune diabetes model; Immunol. Rev. 2000:173, 109-119
14. Saito H., Kranz D.M., Takagaki Y., Hayday A.C. et al.; Complete primary structure of a heterodimeric T-cell receptor deduced from cDNA sequences; Nature 1984:309, 757-762
15. Brenner M.B., McLean J., Dialynas D.P., Strominger J.L. et al.; Identification of a putative second T-cell receptor; Nature 1986:322, 145-149
16. Alvarez A.J., Endsley J.J., Werling D. et al.; WC1(+) gammadelta T cells indirectly regulate chemokine production during mycobacterium bovis infection in SCID-bo mice; Transbound Emerg Dis. 2009:56, 275-284
17. Champagne E.; γδ T cell receptor ligands and modes of antigen recognition; Arch. Immunol. Ther. Exp. (Warsz) 2011:59, 117-137
18. Caccamo N., Dieli F., Wesch D. et al.; Sex-specific phenotypicaland functional differences inperipheral human Vγ9/Vδ2 T cells.; J. Leukoc. Biol. 2006:79, 663-666
19. French J.D., Roark C.L., Born W.B., O’Brien R.L.; γδ T cell homeostasis is established in competition with αβ T cells and NK cells; PNAS 2005:102, 14741-14746
20. Porcelli S.A., Morita C.T., Modlin R.L.; T cell recognition of nonpeptide antigens; Curr. Opin. Immunol. 1996:8, 510-516
21. Jakóbisiak M., Makowski M.; Receptory limfoctów T wiążące antygen. W: Immunologia. M. Jakóbisiak; Wydawnictwo Naukowe PWN Warszawa 2010, 4, 48-53
22. Sikora M., Sinkorova J., Holtmeier W.; Development of cd thymocyte subsets during prenatal and postnatal ontogeny; Immunology 2005:115, 544-555
23. Akirav E.M., Ruddle N.H., Herold K.C.; The role of AIRE in human autoimmune disease; Nat. Rev. Endocrinol. 2011:7, 25-33
24. Lehner T.; Special regulatory T cell review: The resurgence of the concept of contrasuppression in immunoregulation; Immunology 2008:123, 40-44
25. Newton D.J., Andrew E.M., Dalton J.E. et al.; Identification of novel γδ T-cell subsets following bacterial infection in the absence of V1+ T cells: homeostatic control of γδ T-cell responses to pathogen infection by V1+ T cells; Infection and Immunity 2006:2, 1097-1105
26. Hirsh M.I., Junger W.G.; Roles of heat shock proteins and gamma delta T cells in inflammation; Am. J. Respir. Cell. Mol. Biol. 2008:39, 509-513
27. Bartkowiak J., Błoński J.; Aktywność przeciwnowotworowa limfocytów T gamma-delta; Post. Hig. Med. Dośw. 2000:54, 35-51
28. Matsushima A., Ogura H., Fujita K. et al.; Early activation of gammadelta T lymphocytes in patients with severe systemic inflammatory response syndrome; Shock 2004:22, 11-15
29. Sowińska E., Usnarska-Zubkiewicz L., Kuliczkowski K.; Estimation of gamma-delta-1 T cell receptor (γδ-1 TCR) expression in bone marrow lymphocyte population in lymphoma malignum and multiple myeloma; Farmacological Reports 2007:59, 261-268
30. Komano H.; Homeostatic regulation of intestinal epithelia by intraepithelial gamma-delta T cells; Proc. Natl. Acad. Sci. USA 1995:92, 6147-6151
31. Harrison L.C., Dempsey-Collier M., Kramer D.R., Takahashi K.; Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes; J. Exp. Med. 1996:184, 2167-2174
32. Aspord C., Thivolet C.; Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice; Clin. Exp. Immunol. 2002:130, 204-211
33. Han G., Wang R., Chen G., Wang J. et al.; Interleukin-17-producing gammadelta+ T cells protect NOD mice from type 1 diabetes through a mechanism involving transforming growth factor-beta; Immunology 2010:129, 197-206
34. Zhang L., Jin N., Nakayama M. et al.; Gamma delta T cell receptors confer autonomous responsiveness to the insulin peptide B:9–231; J. Autoimmun. 2010:34, 478-484
35. Szczepanik M.; Regulacja reakcji nadwrażliwości kontaktowej przez supresyjne limfocyty T γδ; Folia Medica Cracoviensia 1998:XXXIX, 1-2, 5-32
36. Hoffmann J.C., Pawlowski N.N., Grollich K. et al.; Gammadelta T lymphocytes: a new type of regulatory T cells suppressing murine 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis; Int. J. Colorectal. Dis. 2008:23, 909-920
37. Nanno M., Shiohara T., Yamamoto H. et al.; γδ T cells: firefighters or fire boosters in the front lines of inflammatory responses; Immunol. Rev. 2007:215, 103-113
38. Antvorskov J.C., Fundova P., Buschard K., Funda D.P.; Impact of dietary gluten on regulatory T cells and Th17 cells in BALB/c mice; PLoS One 2012:7, e33315
39. Goldrath A.W., Barber L., Chen K.E., Alters S.E.; Differences in adhesion markers, activation markers, and TcR in islet infiltrating vs. peripheral lymphocytes in the NOD mouse; J. Autoimmun. 1995:8, 209-220
40. Krętowski A., Myśliwiec J., Szelachowska M. et al.; Gamma-delta T-cells alterations in the peripheral blood of high risk diabetes type 1 subjects with subclinical pancreatic beta-cells impairment; Immunol. Lett. 1999:68, 289-293
41. Kretowski A., Myśliwiec J., Kinalska I.; Abnormal distribution of gamma delta T lymphocytes in Graves’ disease and insulin-dependent diabetes type 1; Arch. Immunol. Ther. Exp. (Warsz) 2000:48, 39-42
42. Lang F.P., Pollock B.H., Riley W.J. et al.; The temporal association between gamma delta T cells and the natural history of insulin-dependent diabetes; J. Autoimmun. 1993:6, 107-119
43. Lang F.P., Schatz D.A., Pollock B.H. et al.; Increased T lymphocytes bearing the gamma-delta T cell receptor in subjects at high risk for insulin dependent diabetes; J. Autoimmun. 1991:4, 925-933
44. Roura-Mir I.C., Alcalde L., Vargas F., Tolosa E. et al.; γδ Lymphocytes in endocrine autoimmunity: evidence of expansion in Graves’ disease but not in type 1 diabetes; Clin. Exp. Immunol. 1993:92, 288-295
45. Gyarmati J., Szekeres-Barthó J., Fischer B., Soltész G.; Fetal type lymphocytes in insulin dependent diabetes mellitus; Autoimmunity 1999:30, 63-69
46. Jacobs M.R., Haynes B.F.; Increase in TCR gamma delta T lymphocytes in synovia from rheumatoid arthritis patients with active synovitis; J. Clin. Immunol. 1992:12, 130-138
47. Giacomelli R.; Circulating Vdelta+1 cells are activated and accumulate in the skin of systemic sclerosis patients; Arthritis Rheum. 1998:41, 327-334
48. Olive C., Gatenby P.A., Serjeantson S.W.; Restricted junctional diversity of Tcell receptor delta gene rearrangements expressed in systemic lupus erythematosus (SLE) patients; Clin. Exp. Immunol. 1994:97, 430-438
49. Volc-Platzer B., Anegg B., Milota S. et al.; Accumulation of gamma delta T cells in chronic cutaneous lupus e rythematosus; J. Invest. Dermatol. 1993:100, 84S-91S
50. Zocchi M.R., Ferrarini M., Migone N., Casorati G.; T-cell receptor V delta gene usage by tumor reactive gamma delta T lymphocytes infiltrating human lung cancer; Immunology 1994:81, 234-239
51. Kuriyama Y., Kawanishi Y., Otawa M. et al.; Circulating and tumor-infiltrating gamma delta T cells in patients with B-cell lymphomas; Leuk. Lymphoma 2000:39, 321-327
52. Zheng B.J., Ng S.P., Chua D.T., Sham J.S. et al.; Peripheral gamma delta T-cell deficit in nasopharyngeal carcinoma; Int. J. Cancer., 2002:99, 213-217
53. Groh V., Rhinehart R., Secrist H. et al.; Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB; Proc. Natl. Acad. Sci. USA 1999:96, 6879-6884








