Endokrynol. Ped. 12/2013;3(44):77-84
DOI: 10.18544/EP-01.12.03.1460
Zespół Rohhad
Klinika Endokrynologii i Diabetologii, Instytut Pomnik Centrum Zdrowia Dziecka w Warszawie
Słowa kluczowe: zespół ROHHAD, otyłość, centralna hipowentylacja, dysfunkcja podwzgórza
Streszczenie
ROHHAD (Rapid-onset Obesity, Hypoventilation, Hypothalamic, Autonomic Dysfunction) to rzadko występujący zespół charakteryzujący się szybko narastającą otyłością o wczesnym początku, dysfunkcją podwzgórza, hipowentylacją oraz zaburzeniami układu autonomicznego. Etiologia zespołu pozostaje nieznana, do hipotez należy tło autoimmunologiczne, paranowotworowe, uwarunkowanie genetyczne czy też modyfikacja epigenetyczna. Materiał i metody. W pracy poddano analizie przypadki chorych opisane w piśmiennictwie, w tym jeden przypadek własny autorów. Przedstawiono spektrum objawów klinicznych zespołu oraz częstość ich występowania, obecnie dostępne metody diagnostyczne i terapeutyczne. Wnioski. W przypadku diagnostyki różnicowej otyłości należy pamiętać o zespole ROHHAD. Brak rozpoznania, a co za tym idzie leczenia, stwarza zagrożenie życia pacjenta w mechanizmie zatrzymania czynności układu oddechowego i krążenia. Pacjenci z zespołem ROHHAD powinni pozostawać pod opieką zespołu wielospecjalistycznego. Poznanie etiologii zespołu być może pozwoli na opracowanie testu diagnostycznego oraz wprowadzenie leczenia przyczynowego
Wstęp
ROHHAD (Rapid-onset Obesity, Hypoventilation, Hypothalamic, Autonomic Dysfunction) to rzadko występujący zespół, który charakteryzują otyłość o wczesnym początku, dysfunkcja podwzgórza, hipowentylacja oraz zaburzenia układu autonomicznego. Pierwszy opis objawów klinicznych odpowiadający zespołowi pochodzi z 1965 r. W 2000 r. E. S. Katz i wsp. w czasopiśmie pulmonologicznym (Pediatric Pulmonology) opisali zespół, który nazwano LO-CHS/HD (late onset central hypoventilation/hypothalamus dysfunction) [1]. Autorzy opisali 11 przypadków tego zespołu (1 własny oraz 10 pochodzących z piśmiennictwa). Od 2000 do 2007 r. funkcjonował pod nazwą LO-CHS/HD late onset central hypoventilation/hypothalamus dysfunction (centralna hipowentylacja o późnym początku z dysfunkcją podwzgórza). Dopiero w 2007 r. zespół został nazwany ROHHAD przez Ize-Ludlowa i wsp., którzy opisali 15 pacjentów prezentujących wymienione cechy kliniczne [2]. Dotychczas opisano 76 przypadków, ale wydaje się, że liczba chorych jest zaniżona, zespół nie jest aż tak rzadki, jak wynika to z danych piśmiennictwa [1–12].
ROHHAD to choroba, która nierozpoznana i nieleczona zagraża utratą życia w mechanizmie zatrzymania czynności układu oddechowego i krążenia. Ustalono następujące kryteria rozpoznania ROHHAD: centralne zaburzenia wentylacji >1 roku życia oraz co najmniej jeden z następujących objawów: szybko narastająca otyłość, hiperprolaktynemia, zaburzenia gospodarki wodno-elektrolitowej, wtórna niedoczynność tarczycy, somatotropinowa niedoczynność przysadki, wtórna niedoczynność nadnerczy, opóźnione dojrzewanie płciowe [2].
Bardzo charakterystyczne są objawy wynikające z dysfunkcji podwzgórza. Odgrywa ono rolę homeostatyczną w regulacji hormonalnej, autonomicznej oraz krążeniowo-oddechowej organizmu. Jest głównym centrum autonomicznej kontroli regulacji temperatury, pragnienia, głodu, nastroju, snu, reakcji źrenic na światło. Pierwszym oraz najbardziej charakterystycznym objawem wynikającym z dysfunkcji podwzgórza jest hiperfagia z szybko narastającą otyłością. W zdecydowanej większości przypadków pojawia się ona ok. 2–4 roku życia, ale opisywano również występowanie otyłości później, tj. do ok. 10 roku życia. Przyrost masy ciała jest zwykle gwałtowny i bardzo duży, wynosi ok. 10–15 kg w ciągu 6–12 miesięcy. Następnie po kilku miesiącach lub latach pojawiają się inne objawy niedoczynności układu podwzgórzowo-przysadkowego, najczęściej jest to niedobór hormonu wzrostu (GH), rzadziej wtórna niedoczynność tarczycy i nadnerczy, hipogonadyzm hipogonadotropowy. Bardzo rzadko rozpoznaje się przedwczesne dojrzewanie płciowe centralne. Kolejno pojawiają się objawy ze strony układu autonomicznego, a w końcu najbardziej niebezpieczny z objawów, nieleczony mogący prowadzić do śmierci, czyli centralna hipowentylacja.
Oprócz wspomnianych objawów u 30–50% pacjentów w różnej sekwencji czasowej w stosunku do początku choroby występują guzy pochodzące z grzebieni nerwowych, tj. neuroblastoma, ganglioneuroblastoma, ganglioneuroma. Badania obrazowe mózgu- CT/MRI nie wykazują żadnych nieprawidłowości w okolicy podzwgórzowo-przysadkowej ani też w innych okolicach centralnego układu nerwowego. Podobnie w badaniach autopsyjnych mózgu nie stwierdza się u większości chorych żadnej patologii. Tylko u jednego pacjenta z zespołem ROHHAD Ouvrier, Nunn i wsp. opisali nacieki limfo- i histiocytarne w obrębie podwzgórza [18, 19]. U tego chorego wykazano również obecność ganglioneuroblastoma. Opóźnione rozpoznanie zespołu może doprowadzić w wyniku niedotlenienia mózgu do zaburzeń funkcji poznawczych, padaczki, zaburzeń emocji i zachowania, opóźnienia rozwoju psychoruchowego.
Etiologia
Etiologia zespołu ROHHAD nie została dotychczas poznana. Przebadano do chwili obecnej kilka genów kandydatów [13]. Nie wykazano mutacji genu homeotycznego PHOX2B w locus 4p12, która jest mutacją charakterystyczną dla zespołu wrodzonej ośrodkowej hipowentylacji (CCHS, congenital central hypoventilation syndrome). CCHS to choroba znana również pod nazwą klątwa Ondyny, a jej częstość jest szacowana na 1:200 000 żywych urodzeń [9, 14]. CCHS rożni się od ROHHAD tym, że objawy ze strony układu oddechowego w CCHS, pod postacią niewydolności oddechowej z upośledzoną odpowiedzią na hiperkapnię i hipoksemię, występują głównie podczas snu i pojawiają się bardzo wcześnie, bo już w okresie noworodkowym. Natomiast nie powstają tu zaburzenia hormonalne. U części pacjentów, podobnie jak w ROHHAD, pojawiają się objawy dysfunkcji autonomicznego układu nerwowego, tj. zaburzenia motoryki przewodu pokarmowego, zaburzenia rytmu serca, nieprawidłowa reakcja źrenic na światło, zlewne poty, a także zaburzenia termoregulacji. Ponadto u ok.
14% pacjentów z CCHS występuje choroba Hirschprunga. W obydwu zespołach oprócz zaburzeń oddychania, dysfunkcji układu autonomicznego występują też guzy wywodzące się z grzebieni nerwowych, co mogłoby wskazywać na ich wspólną patofizjologię.
Badano również inne geny, tj. BDNF (brain-derived neurothrophic factor) i gen kodujący receptor TRKB, czyli NTRK2, poza tym takie geny, jak: HTR1A, OTP, PACAP, ASCLI, NECDIN. Wszystkie te geny są zaangażowane w rozwój układu autonomicznego, podwzgórza i systemu neuroendokrynnego. Dotychczas nie wykazano mutacji w obrębie wymienionych genów analizowanych u pacjentów z klinicznie rozpoznanym ROHHAD.
Oprócz czynników genetycznych w etiopatogenezie ROHHAD bierze się również pod uwagę czynniki autoimmunologiczne [15]. Przypuszcza się, że u osoby podatnej genetycznie nieznany czynnik środowiskowy powoduje pojawianie się przeciwciał reagujących z własnymi tkankami. Jednak do chwili obecnej nie wykazano obecności swoistych przeciwciał ani w surowicy, ani też w płynie mózgowo-rdzeniowym u chorych z ROHHAD.
W etiologii zespołu rozważa się też możliwość, iż jest to zespół paranowotworowy, ponieważ u ok. 30–50% pacjentów występują guzy wywodzące się ze zwojów autonomicznych [16–18]. Zespół paranowotworowy to degeneracyjny zespół dotyczący centralnego i obwodowego układu nerwowego, który rozwija się u chorego z nowotworem, a nie jest związany z bezpośrednią inwazją przez guz. Zespół paranowotworowy jest rodzajem autoimmunologicznej odpowiedzi na obecność guza i może być skutecznie leczony terapią immunosupresyjną i/lub usunięciem guza. Zespół mioklonii i opsoklonii oraz ataksja móżdżkowa są znanymi przykładami zespołu paranowotworowego, który spowodowany jest obecnością guza wywodzącego się z grzebieni nerwowych i występuje u 4% pacjentów z tego typu nowotworem. Przeciwko tej hipotezie przemawia fakt, iż guzy u pacjentów z ROHHAD występują tylko u połowy chorych. Poza tym pojawiają się w różnej sekwencji czasowej w stosunku do innych objawów, czasami nawet po 9–14 latach po pojawieniu się pierwszych objawów, ponadto objawy nie cofają się po usunięciu guza oraz po leczeniu immunosupresyjnym. Istnieje pogląd, że być może guzy występują jako pierwszy objaw choroby i są obecne u wszystkich pacjentów, a u części z nich następuje ich samoistna inwolucja. U pacjentów z ROHHAD nie znajdowano autoprzeciwciał (anty-Hu oraz innych przeciwciał przeciwneuronalnych) ani w surowicy, ani też w płynie mózgowo-rdzeniowym, które mogą reagować krzyżowo z komórkami nerwowymi OUN, co jest charakterystyczne dla zespołu paranowotworowego towarzyszącego guzom typu neuroblastoma.
Opisano także przypadek zespołu ROHHAD u jednego z bliźniąt jednojajowych, co dowodzi, że etiologia zespołu nie jest uwarunkowana wyłącznie genetycznie [4]. Możliwe jest dziedziczenie predyspozycji genetycznej, a do zachorowania konieczne są także czynniki środowiskowe lub występuje tu modyfikacja epigenetyczna, jak w przypadku zespołu Pradera-Williego czy zespołu Retta.
Badania niezbędne do ustalenia etiologii zespołu pozwolą wyjaśnić, czy mamy do czynienia z zespołem autoimmunologicznym, paranowotworowym, genetycznym, czy też modyfikacją epigenetyczną. Dotychczasowe wyniki badań nie dały na to pytanie odpowiedzi. Wykrycie testu diagnostycznego, i co za tym idzie rozpoczęcie leczenia, przyczyniłoby się do zmniejszenia chorobowości i śmiertelności w tej grupie chorych oraz poprawy jakości ich życia.
Zmiany nowotworowe
U 30–50% pacjentów z ROHHAD występują guzy wywodzące się z komórek grzebieni nerwowych, nowotwory dotyczą zwojów układu autonomicznego [1–3]. Pod względem histopatologicznym wyróżnia się 3 typy guzów: neuroblastoma, ganglioneuroblastoma i ganglioneuroma. Ten ostatni to najczęściej guz łagodny. W ok. 35% przypadków wydziela katecholaminy (VMA lub HVA). Najczęściej jest zlokalizowany w śródpiersiu tylnym (41,5%), rzadziej w przestrzeni zaotrzewnowej (37,5%), tylko w 21% przypadków w nadnerczach. Ganglioneuroblastoma i neuroblastoma to guzy o pośrednim stopniu złośliwości i guzy złośliwe. W większości przypadków, bo aż u 90–95%, wydzielają katecholaminy. W odróżnieniu od ganglioneuroma najczęściej lokalizują się w rdzeniu nadnerczy (35%), rzadziej w przestrzeni zaotrzewnowej (30%) czy też w śródpiersiu tylnym (20%). Guzy mogą pojawiać się w różnym czasie od pierwszych objawów zespołu, ale najczęściej jako ostatni element choroby, opisywano ich wykrycie nawet po 9–14 latach od zachorowania. W tego typu nowotworach stosuje się leczenie operacyjne, w przypadkach złośliwych leczenie jest uzupełnione chemio- i radioterapią. Ze względu na możliwość występowania tego typu guzów u pacjentów z ROHHAD konieczna jest okresowa diagnostyka obrazowa klatki piersiowej i jamy brzusznej. Zalecane badania obrazowe klatki piersiowej i jamy brzusznej powinny być wykonywane co 12–18 miesięcy, a po 10 latach od rozpoznania co 2 lata.
Objawy kliniczne zespołu ROHHAD
I. Objawy podwzgórzowe:
• hiperfagia i narastająca otyłość: początek 2–4 rok życia, • somatotropinowa niedoczynność przysadki, • wtórna niedoczynność tarczycy, • wtórna niedoczynność nadnerczy, • hipogonadyzm hipogonadotropowy, • przedwczesne dojrzewanie płciowe, • hiperprolaktynemia, • zaburzenia gospodarki wodno-elektrolitowej: hipernatremia, hiponatremia, moczówka prosta (całkowita i częściowa), moczówka prosta z adypsją, SIADH, • zaburzenia termoregulacji: hipotermia, hipotermia, poikilotermia, • nadmierna senność
II. Centralna i obwodowa hipowentylacja:
• zespół bezdechów nocnych, • pęcherzykowa hipowentylacja, • wzrost zużycia tlenu, pułapka powietrzna, zmniejszona wrażliwość na dwutlenek węgla, zmieniony mechanizm oddechowy, uszkodzenie mięśni oddechowych, • epizody sinicy, • zatrzymanie krążenia i oddychania
III. Objawy autonomiczne:
• rozszerzone źrenice, nieprawidłowa reakcja źrenic na światło, • zez, • nadmierna potliwość, brak wydzielania potu i łez, • zaburzenia motoryki przewodu pokarmowego, • bradykardia, • zimne dłonie i stopy, • obniżona percepcja bólu
IV. Zaburzenia sfery emocji i zachowania:
• labilność emocjonalna, • napady agresji, • obojętność emocjonalna, • objawy depresyjne, • zaburzenia psychotyczne, • zaburzenia obsesyjno-kompulsyjne, • epizody katapleksji, • ataki paniki i lęku, • halucynacje, • zespół Touretta
V. Guzy z grzebieni nerwowych:
• ganglioneuroma, ganglioneuroblastoma, neuroblastoma
VI. Inne:
• drgawki, • padaczka, • opóźniony rozwój psychomotoryczny, regresja rozwoju psychomotorycznego
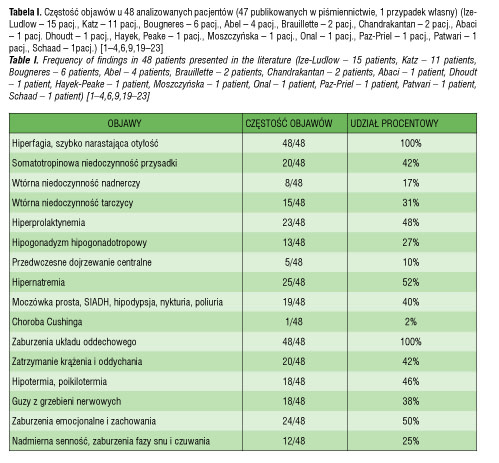
Dwa objawy, które występują u wszystkich pacjentów z zespołem ROHHAD, to hiperfagia z następową otyłością oraz hipowentylacja pęcherzykowa [1–4]. Pierwszą oznaką dysfunkcji podwzgórza jest hiperfagia z szybko narastającą otyłością. Drugim co do częstości objawem, wynikającym z niedoczynności podwzgórzowo-przysadkowej, jest niedobór hormonu wzrostu, występujący u ok. 42% pacjentów. Rzadziej występują wtórna niedoczynność tarczycy, nadnerczy oraz niedoczynność w zakresie gonadotropin. U części pacjentów (10%) rozpoznaje się przedwczesne dojrzewanie płciowe. Stosunkowo częstym objawem, bo występującym aż u połowy pacjentów, jest hiperprolaktynemia. Poziomy prolaktyny wahają się od 30 do 100 ng/ml, śr. ok. 60 ng/ml. Z innych objawów hormonalnych tylko u jednego pacjenta z zespołem ROHHAD stwierdzono ACTH- zależny zespół Cushinga.
U pacjentów z ROHHAD często powstają zaburzenia gospodarki wodno-elektrolitowej, u połowy pacjentów pod postacią hipernatremii. Bardzo często, bo aż u 40% pacjentów, rozpoznaje się różne zaburzenia gospodarki wodno-elektrolitowej, tj. centralną moczówkę prostą całkowitą lub częściową (przejściową lub trwałą), centralną moczówkę prostą z adypsją czy SIADH.
Bardzo charakterystycznym elementem zespołu są zaburzenia układu oddechowego. Występują one, podobnie jak hiperfagia z otyłością, u wszystkich pacjentów z tym rozpoznaniem. U części z nich pojawiają się tylko w nocy, a u innych przez całą dobę. U pacjentów z ROHHAD wykazano najczęściej mieszany typ zaburzeń oddechowych, tj. centralną hipowentylację oraz obturacyjne bezdechy nocne (obstructive sleep apnea). U pacjentów stwierdza się: upośledzoną reakcję na stężenie CO2, hipowentylację pęcherzykową, bezdechy nocne, sinicę, hiperkapnię. Zaburzenia układu oddechowego są niezwykle niebezpieczne. U ok. połowy pacjentów dochodzi do niewydolności oddechowej. Nagłe zgony zdarzają się najczęściej przed postawieniem rozpoznania i występują w nocy. Nie wiadomo, czy są związane z nagłym zatrzymaniem oddychania, czy też wynikają z dysfunkcji układu autonomicznego, a co za tym idzie hipotensji czy też tachy- lub bradyarytmii.
Objawy dysfunkcji układu autonomicznego to także nadmierna lub obniżona potliwość, brak wydzielania łez, zimne dłonie i stopy. U części pacjentów obserwuje się zaburzenia akomodacji pod postacią: szerokich źrenic, braku lub nieprawidłowej reakcji źrenic na światło. Bardzo charakterystycznym i często spotykanym objawem jest obniżona percepcja bólu. Opisywano zaburzenia rytmu serca pod postacią bradykardii, tachykardii, rozkojarzenia przedsionkowo-komorowego, czasem wymagające wszczepienia rozrusznika serca. Objawami stosunkowo często występującymi w tej grupie pacjentów są zaburzenia sfery psychicznej o szerokim spektrum od zaburzeń emocjonalnych i zachowania do zaburzeń psychotycznych. Wśród zaburzeń dotyczących sfery emocjonalnej spotyka się labilność emocjonalną, drażliwość i nadpobudliwość. Mogą pojawiać się również zachowania opozycyjno-buntownicze, napady agresji lub emocjonalna obojętność. Nierzadko możemy obserwować u tych pacjentów zaburzenia koncentracji uwagi i trudności szkolne. Zdarzają się również, choć rzadziej, psychozy, zespoły obsesyjno-kompulsyjne, epizody katapleksji i halucynacje. U pacjentów z ROHHAD opisywano opóźniony rozwój psychomotoryczny lub jego regres. Inne bardzo rzadko występujące objawy dotyczące OUN to drgawki i padaczka.
Różnicowanie
ROHHAD należy różnicować z zespołami charakteryzującymi się wcześnie pojawiającą się otyłością, takimi jak zespół Pradera-Williego, Bardeta-Biedla, niedobór leptyny, mutacja receptora MC4R, mutacja w genie POMC, ale też z chorobą Cushinga. Częstość zespołu Pradera-Williego (PWS) jest szacowana na 1:10 000 do 1:25 000 żywych urodzeń. Najczęściej, w 75% przypadków, występuje delecja fragmentu ojcowskiego długiego ramienia chromosomu 15: 15q11.2-q13, w 24% uniparenteralna disomia matczyna chromosomu 15, w 1%
defekty piętnowania (mikrodelecje w centrum piętnowania), a najrzadziej, bo <1%, translokacje chromosomu ojcowskiego 15q11.2-q13. Do najczęstszych objawów klinicznych zalicza się: otyłość, niedobór wzrostu, hipogonadyzm hipogonadotropowy, rzadziej wtórną niedoczynność nadnerczy. Bardzo charakterystycznymi objawami są też wiotkość mięśniowa, skolioza, opóźnienie rozwoju psychoruchowego i umysłowego, zaburzenia psychiatryczne. Obserwuje się również zaburzenia oddychania w trakcie snu. Do cech wspólnych ROHHAD i PWS należą więc otyłość o wczesnym początku, niedobór wzrostu, wtórna niedoczynność nadnerczy, hipogonadyzm hipogonadotropowy oraz mieszany, czyli centralno-obturacyjny typ zaburzeń oddychania.
Zespół Bardeta-Biedla to wielonarządowy zespół charakteryzujący się, poza otyłością, degeneracją siatkówki, polidaktylią, hipogonadyzmem hipogonadotropowym, trudnościami szkolnymi oraz wadami układu moczowego. Powyższym objawom mogą towarzyszyć inne problemy okulistyczne (zaćma, zez, astygmatyzm), wady uzębienia, cukrzyca, wady serca. Otyłość pojawia się między 2 a 4 rokiem życia. Choroba występuje z częstością od 1:13 500 u mieszkańców Izraela i krajów arabskich do 1:160 000 w Europie. Poznano dotąd 14 genów mogących odpowiadać za rozwój choroby, są to m.in. mutacje w genie BBS 1-8, na chromosomach 2-4,11-16.
Do innych rzadko występujących zespołów wymagających różnicowania z ROHHAD należą niedobór leptyny, mutacja receptora MC4R, mutacja w genie POMC czy też choroba Cushinga.
Leczenie
Terapia zespołu polega na substytucyjnym leczeniu hormonalnym, leczeniu zaburzeń oddechowych oraz usunięciu operacyjnym wykrytych guzów [2]. W przypadku stwierdzenia somatotropinowej niedoczynności przysadki stosuje się preparat hormonu wzrostu. Opisywano dobrą odpowiedź wzrostową po zastosowaniu leczenia hormonem wzrostu, ale terapia ta u większości chorych nie zmienia parametrów związanych z otyłością. W przypadku rozpoznania niedoczynności tarczycy czy nadnerczy konieczna jest substytucja preparatami tyroksyny i hydrokortyzonu. Leczenie hipogonadyzmu hipogonadotropowego polega na terapii hormonami płciowymi. Rzadko rozpoznawane w zespole ROHHAD przedwczesne dojrzewanie płciowe centralne wymaga zastosowania analogu Gn-RH. Leczenie hipowentylacji, w zależności od rodzaju stwierdzanych zaburzeń, polega na zastosowaniu nieinwazyjnej wentylacji dodatnim ciśnieniem przez maskę lub ciągłej mechanicznej wentylacji przez tracheostomię. W razie stwierdzenia komponenty obwodowej hipowentylacji, tj. zaburzeń obturacyjnych, przeprowadza się zabiegi laryngologiczne, takie jak adenotomia, adenotonsillektomia czy plastyka podniebienia. Pacjenci powinni pozostawać pod stałą opieką zespołu specjalistów, w którego skład wchodzą endokrynolog, pulmonolog, anestezjolog, onkolog, a w zależności od specyficznych objawów również lekarze innych specjalności (kardiolog, neurolog, psychiatra, chirurg).
Bardzo ważne są okresowe badania w kierunku guzów typu ganglioneuroma. Badania obrazowe klatki piersiowej i jamy brzusznej powinny być wykonywane co 12–18 miesięcy, przy braku zmian w ciągu 10 lat obserwacji badania można wykonywać rzadziej, tj. co dwa lata.
Dotychczas opisano trzech pacjentów, u których stosowano leczenie immunosupresyjne (immunoglobuliny i steroidy, leki immunosupresyjne, preparaty przeciwciał), ale uzyskane wyniki nie były jednoznaczne. Paz-Priel i wsp. opisali pacjenta z objawami klinicznymi sugerującymi zespół ROHHAD, u którego zastosowano cyklofosfamid w dużej dawce z poprawą kliniczną [19]. Nieskuteczne okazało się użycie Encortonu, Rituximabu oraz immunoglobulin. Jednak u tego pacjenta nie obserwowano zaburzeń oddechowych i diagnoza ROHHAD była niepewna. Sirvent i wsp. opisali brak efektu leczenia u dwu pacjentów z ROHHAD po zastosowaniu leczenia immunosupresyjnego [16]. Należy zachować szczególną ostrożność decydując się na leczenie immunosupresyjne u pojedynczych pacjentów. Ppotrzebne są dalsze badania na większej liczbie chorych, prowadzone pod kontrolą ustalonych biomarkerów z krwi i płynu mózgowo-rdzeniowego, w ramach programów klinicznych.
Podsumowanie
W przypadku stwierdzenia otyłości u małego dziecka (po 2 roku życia), szczególnie w dobie narastającej otyłości w całej populacji, także dziecięcej, należy pamiętać o zespole ROHHAD. Każdy pediatra w diagnostyce różnicowej otyłości powinien brać pod uwagę ten zespół. U pacjenta otyłego z towarzyszącymi objawami z układu autonomicznego należy wykonać badania oceniające czynność układu podwzgórzowo-przysadkowego oraz układu oddechowego. W przypadku postawienia rozpoznania na podstawie obrazu klinicznego konieczne są badania obrazowe klatki piersiowej i jamy brzusznej celem wykluczenia obecności zmian nowotworowych wywodzących się z grzebieni nerwowych. Opóźnione rozpoznanie zespołu może prowadzić do postępu choroby, niedotlenienia ośrodkowego układu nerwowego i w konsekwencji do zaburzeń funkcji poznawczych i opóźnienia rozwoju psychoruchowego. Brak diagnozy, a co za tym idzie leczenia, stwarza również zagrożenie życia pacjenta w mechanizmie zatrzymania czynności układu oddechowego i krążenia, co występuje w 50–60% przypadków. Rokowanie znacznie poprawia się przy jak najwcześniejszym zidentyfikowaniu fenotypu zespołu. Objawy pochodzące z wielu narządów wymagają roztoczenia nad pacjentami wielospecjalistycznej opieki. Do zespołu leczącego chorych z zespołem ROHHAD powinni należeć: endokrynolog, pulmonolog, anestezjolog, onkolog. Chociaż spektrum objawów klinicznych zostało dobrze opisane, patogeneza zespołu jest nadal nieznana, a leczenie jest tylko postępowaniem objawowym. Poznanie etiologii zespołu ROHHAD być może pozwoli na opracowanie testu diagnostycznego oraz leczenia przyczynowego, co przyczyni się do zmniejszenia chorobowości i śmiertelności oraz poprawy jakości życia pacjentów.
Piśmiennictwo
1. Katz E.S., McGrath S., Marcus C.L.; Late-onset central hypoventilation with hypothalamic dysfunction: a distinct clinical syndrome; Pediatr. Pulmonol. 2000 Jan:29(1), 62-68
2. Ize-Ludlow D., Gray J.A., Sperling M.A. et al.; Rapid-Onset Obesity With Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation Presenting in Childhood; Pediatrics 2007:120, e179
3. Bougneres P., Pantalone L., Linglart A. et al.; Endocrine Manifestations of the Rapid-Onset Obesity with Hypoventilation, Hypothalamic, Autonomic Dysregulation, and Neural Tumor Syndrome in Childhood; J. Clin. Endocrinol. Metab. 2008:10, 93(10), 3971-3980
4. Patwari P.P., Rand C.M., Berry-Kravis E.M. et al.; Monozygotic twins discordant for ROHHAD phenotype; Pediatrics 2011:128, e000
5. Chew H.B., Ngu L.H., Keng W.T.; Rapid-onset obesity with hypothalamic dysfunction, hypoventilation and autonomic dysregulation (ROHHAD): a case with additional features and review of the literature; BMJ Case Reports 2011
6. Onal H., Ersen A.; A case of late-onset central Hypoventilation syndrome with hypothalamic dysfunction: through a New phenotype; Turk J. Pediatr. 2010:52, 198-202
7. De Pontual L., Trochet D., Caillat-Zucman S. et al.; Delineation of late onset hypoventilation associated with hypothalamic dysfunction syndrome; Pediatr. Res. 08/2008: 64(6), 689-694
8. Lucas-Herald A.K., Davidson M., Davies P. et al.; Two children with rapid onset obesity combined with respiratory and endocrine dysfunction. Do they have ROHHAD?; Arch. Dis. Child. 2012:5, 97(Suppl_1), A119
9. Chandrakantan A., Poulton T.J.; Anesthetic considerations for rapid-onset obesity, hypoventilation, hypothalamic dysfunction, and autonomic dysfunction (ROHHAD) syndrome in children; Pediatric Anesthesia 2013:1, 23(1), 28-32
10. Frank Y., Kravath R.E., Inoue K. et al.; Sleep apnea and hypoventilation syndrome associated with acquired nonprogressive dysautonomia: clinical and pathological studies in a child; Ann. Neurol. 1981 Jul:10(1), 18-27
11. Proulx F., Weber M.L., Collu R. et al.; Hypothalamic dysfunction in child: distinct syndrome?; Eur. J. Pediatr. 1993:152, 526-529
12. Durivage K.S., Winter R.J., Brouillette R.T. et al.; Idiopathic Hypothalamic Dysfunction and Impaired Control of Breathing; Pediatrics 1985:75, 896-898
13. Rand C.M., Patwari P.P., Rodikova E.A. et al.; Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation: analysis of hypothalamic and autonomic candidate genes; Pediatr. Res. 2011 Oct:70(4), 375-378
14. Rohrer T., Trachsel D., Engelcke G. et al.; Congenital central hypoventilation syndrome associated with Hirschsprung’s disease and neuroblastoma: Case of multiple neurocristopathies; Pediatr. Pulmonol. 2002:1,33 (1), 71-76
15. Armangue T., Petit-Pedrol M., Dalmau J.; Autoimmune Encephalitis in Children; J. Child. Neurol. 2012:4, 27(11), 1460-1469
16. Sirvent N., Berard E., Chastagner P. et al.; Hypothalamic Dysfunction associated with a neuroblastoma: Evidence for new paraneoplastic syndrome?; Med. Pediatr. Oncol. 2003 May:40(5), 326-328
17. Nunn K., Ouvrier R., Sprague T. et al.; Idiopathic hypothalamic dysfunction: a paraneoplastic syndrome?; J. Child. Neurol. 1997 Jun:12(4), 276-281
18. Ouvrier R., Nunn K., Sprague et al.; Idiopathic hypothalamic dysfunction: a paraneoplastic syndrome?; Lancet 1995 Nov:11, 346(8985), 1298
19. Abaci A., Catli G., Bayram E. et al.; A case of rapid-onset obesity with hypothalamic dysfunction, hypoventilation, autonomic dysregulation, and neural crest tumor:ROHHADNET syndrome; Endocr, Pract, 2013 Jan-Feb:19(1), e12-6. doi: 10.4158/EP12140.CR
20. Hayek A., Peake G.T.; Hypothalamic adipsia without demonstrable structural leasion; Pediatrics, 1982:70, 275-278
21. Brouillette R.T., Ilbawi M.N., Hunt C.E.; Phrenic nerve pacing in infants and children: A review of experience and report on the usefulness of phrenic nerve stimulation studies; J. Pediatr. 1983:102, 32-39
22. Paz-Priel I., Cooke D.W., Chen A.R.; Cyclophosphamide for Rapid-Onset Obesity, Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation Syndrome; J. Pediatr. 2011 Feb:158(2), 337-339
23. Schaad U., Vassella F., Zuppinger K. et al.; Hypodipsia-hyponatremia syndrome; Helv. Paediatr. Acta 34, 63-76








