Endokrynol. Ped. 11/2012;1(38):71-82
DOI: 10.18544/EP-01.11.01.1374
Zaburzenia gospodarki wodno-sodowej w przebiegu endokrynopatii u dzieci. Część II. Zaburzenia wydzielania i działania mineralokortykosteroidów
1Klinika Endokrynologii Wieku Rozwojowego Uniwersytetu Medycznego w Łodzi, Klinika Endokrynologii i Chorób Metabolicznych Instytutu Centrum Zdrowia Matki Polki w Łodzi
2Klinika Endokrynologii i Chorób Metabolicznych Instytutu Centrum Zdrowia Matki Polki w Łodzi, Klinika Endokrynologii i Chorób Metabolicznych Uniwersytetu Medycznego w Łodzi
Słowa kluczowe: mineralokortykosteroidy, aldosteron, hiperaldosteronizm, zespół utraty soli, pseudohipoaldosteronizm
Streszczenie
Wśród licznych przyczyn nieprawidłowej homeostazy wodno-elektrolitowej u dzieci należy uwzględnić zaburzenia hormonalne prowadzące do nieprawidłowego bilansu wodnego i/lub sodowego. W pierwszej części opracowania przedstawione zostały fizjologiczne mechanizmy regulacji gospodarki wodnej oraz zaburzenia wytwarzania i/lub wydzielania hormonu antydiuretycznego (ADH) – moczówka prosta ośrodkowa, i zespół nieadekwatnego wydzielania ADH oraz stany związane z defektami receptora ADH – moczówka prosta nefrogenna i nerkopochodny zespół nieadekwatnej antydiurezy, a ponadto – hipernatremia neurogenna i zespół mózgowej utraty soli. W drugiej części omówione zostaną zaburzenia gospodarki wodno-elektrolitowej w przebiegu endokrynopatii, związane z nieprawidłowym wydzielaniem mineralokortykosteroidów (MKS). Ponadto przedstawione będą najważniejsze informacje na temat wybranych tubulopatii wymagających różnicowania z pierwotnymi chorobami endokrynologicznymi, a często będących także przyczyną wtórnie zaburzonej sekrecji MKS
Wstęp
Ważną grupę przyczyn zaburzeń gospodarki wodno-sodowej stanowią stany upośledzonej bądź nadmiernej sekrecji mineralokortykosteroidów (MKS), przede wszystkim aldosteronu, bądź stany obniżonej wrażliwości na ten hormon, czy wreszcie interakcje innych hormonów z receptorem mineralokortykosteroidowym (MKS-R). Działanie aldosteronu polega na zwiększaniu wchłaniania zwrotnego jonów sodowych (Na+), a wydalania jonów potasowych (K+) i wodorowych (H+) w dystalnej części nefronu. Nadmierne wydzielanie aldosteronu prowadzi do zatrzymania sodu i – wtórnie – wody w ustroju. Z kolei w przypadku hipoaldosteronizmu dochodzi do hiponatremii z hiperkaliemią i wystąpienia objawów zespołu utraty soli. Należy w tym miejscu zwrócić uwagę na fakt, że zarówno hiper-, jak i hipoaldosteronizm są często stanami wtórnymi, występującymi w przebiegu innych chorób (oraz ich leczenia, gdyż wiele leków modyfikuje sekrecję aldosteronu). U pacjentów z zaburzeniami wydzielania MKS obecne są zazwyczaj także inne objawy nieprawidłowej czynności kory nadnerczy bądź też objawy warunkujących te zaburzenia chorób ogólnoustrojowych. Ze względu na odmienny patomechanizm wymagają one odrębnego postępowania terapeutycznego niż pierwotne zaburzenia wydzielania aldosteronu. W diagnostyce różnicowej tych zaburzeń należy uwzględnić ponadto stany hipoaldosteronizmu rzekomego oraz zespół pozornego nadmiaru MKS.
Fizjologiczne podstawy regulacji gospodarki sodowej
Mineralokortykosteroidy biorą udział w regulacji wydalania Na+ i K+, co ma istotne znaczenie w utrzymaniu prawidłowej objętości płynów ustrojowych. Jak wspomniano wcześniej, pod wpływem aldosteronu zwiększa się wchłanianie zwrotne Na+ oraz wydalanie H+ i K+, przy czym procesy te zachodzą na drodze transportu aktywnego. Warto zauważyć, że K+ jest jedynym jonem, który może być zarówno reabsorbowany, jak i w sposób aktywny wydzielany do światła cewki nerkowej w procesie wymiany jonów. Omawiany proces wymiany jonów wymaga aktywacji cewkowego kanału sodowego (ENaC) i kanału potasowego ROMK na luminalnym (tj. skierowanym do światła cewki) biegunie komórek, a pompy sodowo-potasowej na biegunie podstawnobocznym komórek cewki dystalnej (ryc. 1). Nabłonkowe kanały sodowe zbudowane są z dwu podjednostek alfa, jednej podjednostki beta1 i jednej podjednostki gamma, przy czym każda podjednostka składa się z dwu domen przezbłonowych, jednej domeny pozakomórkowej i jednej domeny cytoplazmatycznej. Kanały te znajdują się nie tylko w komórkach cewki dalszej nefronów, ale także w płucach i w odcinku dystalnym jelita grubego, gdzie również biorą udział w resorpcji sodu i wody. Pompa sodowo-potasowa jest natomiast enzymem o aktywności ATP-azy. Składa się ona z dwu podjednostek alfa i dwu podjednostek beta, tworzących tetramer. Miejsce wiązania ATP znajduje się na podjednostce alfa, do której także – po związaniu jej z ATP – przyłączają się trzy jony Na+, które następnie są transportowane na zewnątrz komórki, czyli „wracają” do osocza. Jednocześnie dochodzi do przyłączenia dwu jonów K+ i ich przeniesienia do wnętrza komórki [1].

Wpływ aldosteronu na równowagę sodowo-potasową polega na zwiększaniu zarówno gęstości i aktywności ATP-azy sodowo-potasowej na błonie podstawnobocznej komórek cewek dystalnych, jak i liczby kanałów ENaC na powierzchni luminalnej tych komórek [2]. Wykazano również, że synteza i egzocytoza ENaC są stymulowane nie tylko przez aldosteron, ale także przez ADH [3, 4]. Warto wspomnieć, że wyniki badań nad ATP-azą sodowo-potasową uhonorowane zostały Nagrodą Nobla w dziedzinie chemii, którą otrzymał w 1997 r. Jens C. Skou. W regulacji gospodarki sodowej istotne znaczenie ma fakt, że kortyzol, który jest wydzielany w znacznie większych ilościach niż aldosteron, ma zdolność wiązania się z MKS-R i jego aktywacji. W tkankach bogatych w MKS-R musi zatem istnieć mechanizm uniemożliwiający to działanie. Polega on na miejscowym przekształcaniu w tych tkankach kortyzolu w kortyzon przez dehydrogenazę 11β-hydroksysteroidową typu 2 (11β-HSD2) (ryc. 2).
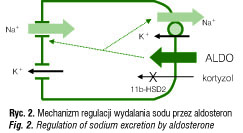
W odróżnieniu od glikokortykosteroidów (GKS) i androgenów, których synteza w korze nadnerczy podlega stymulacji przez ACTH, najważniejszą rolę w pobudzaniu wydzielania MKS odgrywa układ renina–angiotensyna oraz wykazujące działanie synergistyczne jony K+. Pod wpływem reniny, syntetyzowanej w aparacie przykłębkowym nerek, dochodzi do odszczepienia angiotensyny I od cząsteczki wyjściowego substratu – angiotensynogenu. Następnie, pod wpływem konwertazy angiotensyny (ACE), peptyd ten jest przekształcany w angiotensynę II, która bezpośrednio pobudza syntezę i wydzielanie aldosteronu, zwiększając aktywność 18-hydroksylazy i 18-oksydazy – enzymów działających na końcowych etapach biosyntezy aldosteronu w warstwie kłębkowatej kory nadnerczy. Najsilniejszymi bodźcami zwiększającymi sekrecję reniny są: hipowolemia, obniżenie ciśnienia perfuzyjnego nerek, hiponatremia, a także pobudzenie układu współczulnego, podczas gdy hiperwolemia i hipernatremia hamują wytwarzanie reniny.
W regulacji wydalania sodu biorą udział także peptydy sodopędne. Przedsionkowy peptyd sodopędny (ANP) wytwarzany jest w kardiomiocytach pod wpływem hipernatremii i hiperwolemii i gromadzony w prawym przedsionku serca. Wykazuje on działanie hamujące wchłanianie zwrotne jonów Na+, przede wszystkim w kanalikach zbiorczych nefronu, zwiększając w ten sposób natriurezę; jednocześnie czynnik ten zwiększa filtrację nerkową.
Ponadto ANP działa hamująco zarówno na układ renina–angiotensyna–aldosteron (RAA), jak i wydzielanie ADH. Drugim peptydem sodopędnym jest peptyd typu B (BNP), określany niekiedy mianem „mózgowego” (gdyż po raz pierwszy został zidentyfikowany w komórkach mózgu świni) [5]. Jest on również syntetyzowany w kardiomiocytach, ale – odmiennie niż ANP – w obrębie komór, a nie przedsionków serca. Synteza i uwalnianie BNP zwiększają się pod wpływem wzrostu ciśnienia w obrębie jam serca i zwiększonego napięcia miocytów, a mechanizmy regulujące te procesy są bardzo złożone [6]. Działanie BNP polega na zwiększaniu natriurezy, a także filtracji kłębkowej, co prowadzi do zwiększenia diurezy. Powoduje on ponadto rozszerzenie naczyń krwionośnych, a przez to obniżenie ciśnienia tętniczego i zmniejszenie obciążenia wstępnego komór serca. Podobnie jak ANP, wykazuje on również działanie hamujące układ RAA. Opisano także peptyd natriuretyczny typu C, którego działanie polega jednak przede wszystkim na rozszerzaniu naczyń krwionośnych. Peptydy sodopędne mają istotne znaczenie kliniczne przede wszystkim u chorych z niewydolnością serca, u których ich oznaczanie może być istotne w ocenie stopnia zaawansowania choroby [7].
Przedstawione informacje ułatwiają zrozumienie patomechanizmów zaburzeń gospodarki wodno-sodowej oraz nowych kierunków w ich terapii.
Hiperaldosteronizm pierwotny
Mianem hiperaldosteronizmu pierwotnego określa się stany, w których aldosteron wytwarzany jest w zwiększonej ilości w sposób autonomiczny, niezależnie od stymulacji ze strony układu renina–angiotensyna. Konsekwencją nadmiaru aldosteronu jest zwiększone wchłanianie zwrotne Na+ w cewkach nerkowych, co prowadzi do zatrzymania wody w ustroju, a także do zwiększonej ucieczki jonów K+ z moczem (wskutek działania pompy sodowo-potasowej). Konsekwencją zatrzymania sodu i wody jest początkowo hiperwolemia, z upływem czasu dochodzi jednak do zwiększenia diurezy i przywrócenia normowolemii. Zjawisko to związane jest ze zwiększeniem sekrecji ANP. Kliniczną manifestacją hiperaldosteronizmu pierwotnego jest zespół Conna, który charakteryzuje nadciśnienie tętnicze (często o ciężkim przebiegu opornym na leczenie) oraz hipokaliemia. Aldosteron działa także na naczynia krwionośne, powodując włóknienie i przerost ściany tętnic oraz zmniejszenie ich podatności, a także przerost mięśnia sercowego. Dla tych ostatnich zjawisk istotne jest, że oprócz wytwarzania w korze nadnerczy aldosteron syntetyzowany jest także miejscowo w ścianie naczyń i w sercu. Hiperaldosteronizm jest zatem uznanym czynnikiem ryzyka chorób układu sercowo-naczyniowego nie tylko jako przyczyna nadciśnienia tętniczego, ale także ze względu na niekorzystny wpływ na naczynia krwionośne. Istotnym patomechanizmem jego działania okazała się indukcja stresu oksydacyjnego, związana z wtórnymi do hiperaldosteronizmu zaburzeniami gospodarki wapniowej i magnezowej (zwiększone wydalanie Ca2+ i Mg2+, wtórna nadczynność przytarczyc) oraz zaburzeniami homeostazy cynku [8–10]. Według obecnego stanu wiedzy zaburzenia dotyczące układu sercowo-naczyniowego i nerek, a także insulinooporność są – przynajmniej częściowo – związane z działaniem aldosteronu, zarówno pozagenomowym, jak i genomowym, za pośrednictwem MKS-R [11]. Hiperaldosteronizm jest zatem istotnym elementem w patogenezie nie tylko opornego na leczenie nadciśnienia tętniczego, ale także zespołu kardiometabolicznego, chorób układu sercowo-naczyniowego i nerek. Hiperaldosteronizm jest zaburzeniem dotyczącym przede wszystkim osób dorosłych, niemniej należy brać pod uwagę możliwość pojawienia się składowych zespołu metabolicznego już u młodzieży, a nawet u dzieci i ich potencjalny związek ze zwiększonym wydzielaniem aldosteronu. W ostatnich latach ukazało się kilka publikacji poświęconych tym zagadnieniom, jednak ich szczegółowe omówienie wykracza poza ramy niniejszego opracowania.
Przyczyną hiperaldosteronizmu pierwotnego mogą być gruczolaki kory nadnerczy, obustronny przerost kory nadnerczy, rzadziej raki kory nadnerczy i nowotwory o lokalizacji pozanadnerczowej, wytwarzające aldosteron. Choroby te dotyczą przede wszystkim osób dorosłych. Omawiając to zagadnienie w aspekcie pediatrycznym należy zwrócić szczególną uwagę na zagadnienie hiperaldosteronizmu rodzinnego.
W warunkach prawidłowych wytwarzanie aldosteronu jest praktycznie niezależne od działania ACTH. Najważniejszy z enzymów w końcowym etapie tego procesu – syntaza aldosteronu – kodowany jest przez gen CYP11B2. Z kolei 11β-hydroksylaza – enzym przekształcający 11-deoksykortyzol w kortyzol – kodowana jest przez gen CYP11B1, a aktywność tego enzymu podlega regulacji przez ACTH. Oba te geny zlokalizowane są na długim ramieniu chromosomu 8 w odległości zaledwie 30–40 par zasad od siebie [12]. W tych warunkach może dojść do wytworzenia genu chimerycznego, warunkującego zależną od ACTH syntezę aldosteronu w warstwie kłębkowatej kory nadnerczy. Gen ten zawiera sekwencje końca 5’ genu CYP11B1, w tym także region promotorowy tego genu oraz sekwencje końca 3’ genu CYP11B2 [13]. Zaburzenie to określane jest mianem hiperaldosteronizmu rodzinnego typu I (ang. familial hyperaldosteronism type I – FH-I). W leczeniu stosuje się deksametazon, który hamuje wydzielanie ACTH, a przez to – wtórnie – wytwarzanie aldosteronu. Stąd też ta jednostka chorobowa określana jest również jako hiperaldosteronizm poddający się leczeniu GKS (ang. glucocorticoid supressible/remediable hyperaldosteronism). Zaburzenie to dziedziczone jest jako cecha autosomalna dominująca. U większości pacjentów obserwuje się ciężkie nadciśnienie tętnicze już we wczesnych okresach życia, ale w części przypadków nadciśnienie jest łagodne lub nie stwierdza się go w ogóle [14–15]. Opisano także hiperaldosteronizm rodzinny typu 2 (FH-II), którego podłoże genetyczne jest nieznane, a nadmierne wydzielanie aldosteronu w tej chorobie nie jest zależne od ACTH. U pacjentów z rodzin dotkniętych tym zaburzeniem obserwuje się przerost lub obecność gruczolaków kory nadnerczy, a cechy kliniczne, biochemiczne i morfologiczne choroby są takie same, jak w przypadku pierwotnego hiperaldosteronizmu występującego sporadycznie. Rozpoznanie opiera się zatem na stwierdzeniu wysokiego stosunku stężeń aldosteronu do reniny u przynajmniej dwu członków rodziny oraz wykluczeniu obecności genu chimerycznego charakterystycznego dla FH-I [16]. W 2008 r. opisano kolejny występujący rodzinnie wariant hiperaldosteronizmu rodzinnego niepoddającego się leczeniu GKS, o jeszcze niezidentyfikowanym podłożu genetycznym, w którym wykluczono obecność mutacji w obrębie locus genu syntazy aldosteronu oraz szereg innych defektów genetycznych – określany jako typ 3 choroby (FH-III) [17]. Zaburzenie to charakteryzuje się występowaniem od wczesnego dzieciństwa ciężkiego nadciśnienia tętniczego z hipokaliemią oraz szybko postępującym uszkodzeniem narządów wrażliwych na działanie aldosteronu i nie poddaje się leczeniu farmakologicznemu (spironolakton z amiloridem), a jedyną opcją terapeutyczną w tych przypadkach może być obustronna adrenalektomia [17].
W 2011 r. ukazały się doniesienia Mulatero i wsp. [18] oraz Aglony i wsp. [19], które wskazują na możliwość „niedoszacowania” częstości występowania pierwotnego hiperaldosteronizmu rodzinego jako przyczyny nadciśnienia tętniczego u dzieci.
Hiperaldosteronizm wtórny
Przyczyną tego zaburzenia jest długotrwałe, nadmierne pobudzenie układu renina–angiotensyna, prowadzące do zwiększonego wydzielania aldosteronu. Większość chorych to osoby dorosłe z chorobami powodującymi utratę sodu bądź hipowolemię, a więc chorzy z niewydolnością krążenia, chorobami nerek (zwężenie tętnicy nerkowej, zespół nerczycowy), marskością wątroby. Należy brać pod uwagę przedawkowanie leków moczopędnych i przeczyszczających oraz stosowanie preparatów zawierających estrogeny, które nasilają syntezę angiotensynogenu. Rzadką przyczyną hiperaldosteronizmu wtórnego może być obecność guza wydzielającego reninę.
Pozorny nadmiar MKS
Jak wspomniano wcześniej, w warunkach prawidłowych istnieje mechanizm zapobiegający stymulacji MKS-R przez kortyzol. Funkcję taką pełni
11β-HSD2, utleniająca kortyzol do jego nieaktywnego metabolitu – kortyzonu. Enzym ten obecny jest przede wszystkim w tkankach bogatych
w MKS-R. Niedobór 11β-HSD2 może prowadzić do nieprawidłowej, nadmiernej stymulacji MKS-R przez kortyzol (ryc. 3).
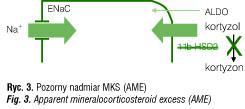
Opisano przypadki mutacji genu kodującego ten enzym, dziedziczonych autosomalnie recesywnie, w których dochodzi do stymulacji MKS-R przez kortyzol w nerkach (a także w innych tkankach: m.in. w gonadach, ośrodkowym układzie nerwowym, łożysku, śliniankach, trzustce) [20] . Zaburzenie to, opisane po raz pierwszy w 1986 r., określane jest mianem pozornego nadmiaru MKS (ang. apparent mineralocorticoid excess – AME) [21]. W ustaleniu rozpoznania istotne jest udokumentowanie podwyższonego stosunku metabolitów kortyzolu (tetrahydrokortyzolu – THF) do metabolitów kortyzonu (tetrahydrokortyzonu – THE), a potwierdzeniem jest wynik badania genetycznego. W niektórych przypadkach stosunek THF:THE jest prawidłowy, co pozwoliło na wyodrębnienie II typu AME (podczas gdy jako typ I kwalifikowane są przypadki z podwyższonym stosunkiem THF:THE) [22]. Zazwyczaj obserwuje się ciężkie nadciśnienie tętnicze z hipokaliemią, występujące już w dzieciństwie z towarzyszącym uszkodzeniem serca, siatkówki i ośrodkowego układu nerwowego [23], ale opisano także przypadki, w których nadciśnienie tętnicze stwierdzone było dopiero w wieku dorosłym i nie towarzyszyły mu zaburzenia elektrolitowe [24]. W terapii wykorzystuje się antagonistów MKS-R (spironolakton) oraz leki hamujące aktywność EnaC (amilorid, triamteren). Należy pamiętać także o możliwości nabytych niedoborów 11β-HSD2, spowodowanych działaniem kwasu glicerytynowego zawartego w lukrecji (Glycyrrhiza glabra), flawonoidów obecnych w soku grejpfrutowym czy karbenoksolonu [25].
Zespoły nadmiaru dezoksykortykosteronu
Dezoksykorytoksteron (DOC) jest jednym z pośrednich metabolitów na szlaku biosyntezy aldosteronu, powstającym z progesteronu wskutek działania 21α-hydroksylazy. Jest on następnie przekształcany w kortykosteron przez 11β-hydroksylazę (P450c11B), a zatem brak lub niedobór tego ostatniego enzymu może być przyczyną z jednej strony niedoboru aldosteronu, a z drugiej – nagromadzenia w nadmiarze DOC. Klinicznie omawiany defekt steroidogenezy manifestuje się jako jedna z postaci wrodzonego przerostu nadnerczy (WPN), którego charakterystyczną cechą (obok typowych objawów wynikających z hiperandrogenizacji) jest rozwój nadciśnienia tętniczego. Zagadnienie to omawiane jest szczegółowo w podręcznikach endokrynologii i w opracowaniach poświęconych WPN, które ukazały się w ostatnich latach w języku polskim, np. w Endokrynologii Polskiej [26, 27] i w Klinice Pediatrycznej [28]. Obraz kliniczny w przypadku niedoboru 11β-hydroksylazy jest bardzo zróżnicowany. Należy mieć na uwadze, że w okresie noworodkowym, zanim dojdzie do nagromadzenia DOC, niedobór aldosteronu może być przyczyną wystąpienia objawów zespołu utraty soli. Nadciśnienie tętnicze z normo- lub hipernatremią i hipokaliemią pojawia się zazwyczaj po trzecim roku życia, ale w niektórych przypadkach może być stwierdzane już w okresie noworodkowym. Nadmierne wytwarzanie DOC odbywa się głównie w warstwie pasmowatej kory nadnerczy, w której zależy ono od wysokiego wydzielania ACTH. W warstwie kłębkowatej proces ten nie jest pobudzany, gdyż podlega on tu pobudzaniu przez reninę, której stężenie w warunkach nadmiaru MKS (w tym przypadku nadmiaru DOC) jest niskie. Pomimo że DOC jest słabym MKS, przy jego nagromadzeniu ujawnia się przewaga działania nadmiaru DOC nad brakiem działania aldosteronu. W terapii stosuje się glikokortykosteroidy, z których u dzieci zalecany jest hydrokortyzon. Należy pamiętać, że po włączeniu leczenia obniża się wydzielanie ACTH, a zatem dochodzi nie tylko do zahamowania syntezy androgenów, ale także do wyraźnego zmniejszenia wytwarzania DOC w warstwie pasmowatej kory nadnerczy. W tej sytuacji wzrasta wydzielanie reniny, która stymuluje wytwarzanie DOC w warstwie kłębuszkowatej kory nadnerczy. Przejściowo w momencie włączenia leczenia hydrokortyzonem może jednak dojść do wystąpienia objawów niedoboru MKS, czyli zespołu utraty soli, wymagających nawet dołączenia do terapii fludrokortyzonu. W leczeniu nadciśnienia tętniczego w przypadkach , w których nie ulega ono normalizacji pod wpływem hydrokortyzonu, stosuje się spironolakton lub amilorid oraz blokery kanału wapniowego, nie są natomiast zalecane inhibitory konwertazy angiotensyny.
Przyczyną wydzielania nadmiernych ilości DOC może być także inny, znacznie rzadszy defekt steroidogenezy nadnerczowej – niedobór 17α-hydroksylazy (a dokładnie enzymu P450c17, posiadającego aktywność 17α-hydroksylazy i 17,20-liazy, działającego w nadnerczach i gonadach). Obraz kliniczny tej postaci WPN wynika ze współistnienia zaburzeń biosyntezy steroidów zarówno w nadnerczach, jak i w gonadach; te ostatnie stanowią przyczynę zaburzeń różnicowania płci (szczegółowe omówienie tych zagadnień można znaleźć we wspomnianych wcześniej pozycjach piśmiennictwa [26–28]). Działanie 17α-hydroksylazy polega na przekształcaniu pregnenolonu w 17α-pregnenolon i progesteronu w 17α-progesteron. W tej sytuacji progesteron nie może być substratem dla biosyntezy kortyzolu czy steroidów płciowych, a jedynym możliwym
szlakiem jego metabolizmu jest przekształcenie w DOC pod wpływem 21α-hydroksylazy. Nadmiar DOC jest przyczyną nadciśnienia tętniczego z hipokaliemią, prowadzi także do zahamowania wydzielania reniny i angiotensyny. Należy zwrócić uwagę na to, że w przypadku niedoboru 17α-hydroksylazy zachowana jest aktywność wszystkich enzymów niezbędnych do biosyntezy aldosteronu i w momencie włączenia leczenia hydrokortyzonem, gdy dojdzie do normalizacji wydzielania reniny, uruchamia się prawidłowe wydzielanie aldosteronu. Trzeba wspomnieć, że w przypadku izolowanego niedoboru 17,20-liazy biosynteza GKS i MKS jest prawidłowa, a zaburzenia dotyczą jedynie wytwarzania steroidów płciowych.
Kolejną przyczyną nadmiaru DOC może być wydzielanie tego hormonu przez nowotwory. Leczenie jest wówczas operacyjne po odpowiednim przygotowaniu pacjenta. Zagadnienie to jest domeną endokrynologii osób dorosłych.
Mutacje receptora MKS
Możliwe jest występowanie mutacji receptora MKS w domenie wiążącej hormon, które sprawiają, że receptor ten może być pobudzany przez inne ligandy. Z praktycznego punktu widzenia istotne znaczenie ma możliwość pobudzania receptora MKS przez progesteron, co ma szczególne znaczenie u kobiet ciężarnych, czy też przez 17-OH-progesteron [29]. Opisano także mechanizm paradoksalnego pobudzenia receptora MKS przez antagonistów aldosteronu, np. spironolakton [30]. W leczeniu stosuje się blokery ENaC – amilorid i triamteren.
Hipoaldosteronizm
Wśród najważniejszych przyczyn hipoaldosteronizmu pierwotnego należy wymienić pierwotną niedoczynność kory nadnerczy o różnym podłożu, wrodzone defekty steroidogenezy nadnerczowej – niedobór 21α-hydroksylazy, niedobór 3β-dehydrogenazy oraz niedobór syntazy aldosteronu, działanie inhibitorów steroidogenezy (np. ketokonazolu czy mitotanu), a także obustronną adrenalektomię. Konsekwencją niedoboru aldosteronu jest zwiększona utrata sodu z moczem, prowadząca do wystąpienia objawów zespołu utraty soli (ryc. 4).
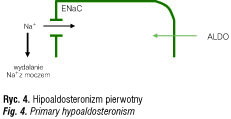
Warto w tym miejscu przypomnieć, że w przypadku wtórnej niedoczynności kory nadnerczy, spowodowanej niedoborem ACTH, wydzielanie aldosteronu jest zazwyczaj prawidłowe, ponieważ jest zależne od pobudzenia układu RAA, a nie od stymulacji przez ACTH.
Do wystąpienia pierwotnej niedoczynności kory nadnerczy (choroby Addisona) dochodzi najczęściej na tle autoimmunologicznym, jakkolwiek należy pamiętać o możliwym związku przyczynowym tej choroby z gruźlicą i zakażeniami grzybiczymi. Objawy kliniczne związane są z niedoborem GKS i MKS, charakterystyczne jest ciemne zabarwienie skóry i błon śluzowych spowodowane nadmiarem melanotropiny (MSH). Szczegółowe omówienie tej jednostki chorobowej wykracza poza ramy obecnego opracowania. Przyczyną ostrej niewydolności kory nadnerczy może być ich uszkodzenie w przebiegu obustronnego do nich krwawienia. Sytuacja taka może wystąpić w okresie noworodkowym lub nawet prenatalnym, a wśród czynników przyczynowych wymienia się uraz okołoporodowy, niedotlenienie, zakażenia wewnątrzmaciczne i zaburzenia koagulologiczne. Przypadki takie prezentowane były m.in. podczas XV Sympozjum PTED w 2006 r. [31].Należy pamiętać o ryzyku wystąpienia ostrej niewydolności nadnerczy w przebiegu posocznicy, czyli zespołu Waterhouse’a-Friedrichsena. U chłopców jedną z rzadszych przyczyn niewydolności kory nadnerczy może być także adrenoleukodystrofia – choroba o dziedziczeniu sprzężonym z chromosomem X, spowodowana mutacją genu ABCD1 prowadzącą do zaburzeń β-oksydacji długołańcuchowych kwasów tłuszczowych (VLCFA) w peroksysomach i w konsekwencji – do nagromadzenia VLCFA w różnych tkankach. Gromadzące się w nadmiarze VLCFA w korze nadnerczy łączą się z cholesterolem, tworząc oporne na działanie hydrolaz estry, co prowadzi do ograniczenia biosyntezy hormonów steroidowych wskutek niedostępności ich prekursora, jakim jest cholesterol. Przedstawione zaburzenia metaboliczne są także przyczyną postępującej demielinizacji w obrębie OUN z jej konsekwencjami neurologicznymi [32]. Szczegółowy opis adrenoleukodystrofii, ze szczególnym uwzględnieniem jej charakterystyki endokrynologicznej, przedstawili w Endokrynologii Pediatrycznej w 2004 r. Fichna i wsp. [33, 34]. Warto zwrócić uwagę, że w przypadku innej jednostki chorobowej o tym samym mechanizmie dziedziczenia – wrodzonej hipoplazji nadnerczy, związanej z mutacją genu DAX1 – dochodzi do wybiórczego niedoboru GKS i androgenów, podczas gdy wytwarzanie aldosteronu jest zachowane [35].
W terapii pacjentów z hipoaldosteronizmem pierwotnym podstawowe zastosowanie ma fludrokortyzon. Z praktycznego punktu widzenia należy pamiętać, że również hydrokortyzon w wysokich dawkach wykazuje działanie na receptor MKS, co może mieć istotne znaczenie u pacjentów w stanie zagrożenia życia, zwłaszcza że fludrokortyzon dostępny jest w Polsce jedynie w formie tabletek (Cortineff) oraz preparatów do stosowania zewnętrznego, a nie ma postaci tego leku do stosowania pozajelitowego.
Przyczyną hipoaldosteronizmu wtórnego jest zahamowanie czynności osi RAA. W patomechanizmie tych zaburzeń należy uwzględnić działanie wielu leków ukierunkowane właśnie na obniżenie biosyntezy aldosteronu (inhibitory konwertazy angiotensyny) czy też leków hamujących wydzielanie reniny, stosowanych z innych wskazań (niesteroidowe leki przeciwzapalne, cyklosporyna, β-blokery) oraz heparyny. Objawy hipoaldosteronizmu mogą wystąpić także w przypadku przedawkowania blokerów receptora aldosteronu.
Pseudohipoaldosteronizm
Mianem pseudohipoaldosteronizmu (PHA) określa się stany, w których dochodzi do niewrażliwości tkanek docelowych na działanie MKS. Przyczyną PHA mogą być defekty genetyczne, i wówczas ma on charakter trwały, bądź też różne patologie w obrębie układu moczowego i wówczas zjawisko to jest zazwyczaj przejściowe.
Wśród postaci uwarunkowanych genetycznie wyróżnia się PHA typu 1 (PHA1), w którym mutacje dotyczą MKS-R (postać nerkowa) lub kanału sodowego ENaC (postać wielonarządowa) (ryc. 5), oraz PHA typu 2 (PHA2), którego przyczyną są mutacje dotyczące rodziny kinaz serynowo-treoninowych: WNK1 i WNK4. Kinazy te regulują aktywność kotransportera sodowo-chlorkowego (NCC) i kanału potasowego ROMK [36].
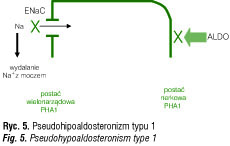
Pseudohipoaldosteronizm typu 1 może mieć postać nerkową i postać wielonarządową. Postać nerkowa PHA1, związana z mutacjami genu hMR kodującego typ I receptora MKS, dziedziczy się autosomalnie recesywnie. Wymaga ona różnicowania z częściej występującym przejściowym PHA1, spowodowanym różnymi patologiami układu moczowego (wady rozwojowe, uropatia zaporowa, infekcje układu moczowego, amyloidoza), które zaburzają wydalanie sodu przez nerki [37, 38]. Pierwszy opis PHA1 u niemowlęcia pochodzi z roku 1958 [39].
W Polsce przypadki niemowląt z zespołem utraty soli w przebiegu PHA1 przedstawili w 2007 r. Szalecki i wsp. [37]. U noworodków z PHA1 stwierdza się objawy zespołu utraty soli z odwodnieniem, hiponatremią i hiperkaliemią oraz kwasicą metaboliczną. Dodatkową wskazówką ułatwiającą rozpoznanie jest brak odpowiedzi na leczenie MKS. Stwierdza się wysokie stężenie aldosteronu w osoczu i podwyższoną w nim aktywność reninową; rozpoznanie można potwierdzić także na podstawie wyniku badania profilu steroidów w moczu [38]. Przebieg kliniczny choroby może być bardzo zróżnicowany, łącznie z postaciami bezobjawowymi, możliwe są także okresy remisji. W terapii stosuje się suplementację chlorku sodowego (NaCl) lub wodorowęglanu sodowego (NaHCO3). Zazwyczaj około drugiego roku życia dochodzi do samoistnej normalizacji stężeń elektrolitów w osoczu, ale mogą się utrzymywać podwyższone poziomy aldosteronu [37]. W przejściowym PHA1 oporność cewek nerkowych na aldosteron ustępuje zwykle po wyleczeniu pierwotnej przyczyny tego stanu (np. antybiotykoterapia zakażeń układu moczowego, leczenie chirurgiczne nefropatii zaporowej), ale niekiedy dochodzi do przetrwałej oporności na aldosteron u pacjentów po zabiegach chirurgicznych. W 2011 r. przedstawiono także przypadek przejściowego PHA1 u chłopca z zaburzeniami rozwoju zewnętrznych narządów płciowych, którego obraz kliniczny sugerował rozpoznanie WPN, jednak poprawę stanu dziecka uzyskano dopiero w wyniku dożylnej suplementacji NaCl [40]. Postać wielonarządowa PHA1, związana z mutacjami ENaC, jest dziedziczona autosomalnie recesywnie. Ze względu na to, że kanały ENaC obecne są nie tylko w nerkach, ale także w innych tkankach epitelialnych, zaburzenia mogą dotyczyć także innych narządów – przede wszystkim płuc, gdzie w związku z zaburzoną absorpcją sodu i wody obserwuje się zaleganie zwiększonej ilości płynu w drogach oddechowych, ale także ślinianek i skóry (stwierdza się zwiększone stężenie sodu w ślinie i w pocie) oraz błony śluzowej jelita grubego [41, 42]. Przebieg wielonarządowej postaci PHA1 jest na ogół cięższy i nie obserwuje się samoistnej poprawy w okresie wczesnego dzieciństwa [36].
Pseudohipoaldosteronizm typu 2 związany jest z pierwotnym defektem cewkowym, prowadzącym do nadmiernej reabsorpcji NaCl ze zwiększeniem objętości płynu pozakomórkowego. Choroba jest zatem jedną z tubulopatii, w których przebiegu obserwuje się zaburzenia gospodarki sodowej, bardziej znana jest pod nazwą zespołu Gordona. Patomechanizm i obraz kliniczny, a także sposób postępowania w przypadku PAH1 i PAH2 są zatem całkowicie odmienne.
Zaburzenia gospodarki sodowej w przebiegu tubulopatii
Zespół Gordona (czyli PAH2) spowodowany jest nadmierną reabsorpcją chlorków z towarzyszącym zwiększonym wchłanianiem zwrotnym sodu
i wody, co prowadzi do wtórnej supresji wydzielania reniny i aldosteronu. Manifestuje się nadciśnieniem (u dzieci ciśnienie tętnicze krwi może być
prawidłowe) z hiperkaliemią i łagodną kwasicą metaboliczną. Hiperkaliemia w tym zespole związana jest ze zmniejszonym dopływem sodu do cewki dystalnej, prowadzącym do zmniejszenia jego wymiany na potas. Choroba dziedziczy się autosomalnie recesywnie. Leczenie polega na ograniczeniu podaży sodu w diecie oraz stosowaniu hydrochlorotiazydu. Pierwsze przypadki PHA2 opisano w Australii w 1964 r. [43] i w 1970 r. [44].
Zespół Gitelmana jest „kliniczną odwrotnością” zespołu Gordona [45]. Związany jest z mutacjami genów kodujących kotransporter sodowo-chlorowy wrażliwy na tiazydy oraz kanały magnezowe w cewce dystalnej. Obraz kliniczny odpowiada zaburzeniom obserwowanym w przypadku przedawkowania diuretyków tiazydowych. Pacjenci mają obniżone ciśnienie tętnicze, skarżą się na ogólne osłabienie, obniżenie siły mięśniowej i skurcze mięśni, a w części przypadków obserwuje się porażenie hipokaliemiczne. Przebieg kliniczny choroby jest bardzo zróżnicowany – od postaci bezobjawowych aż do ciężkich objawów ze strony układu nerwowo-mięśniowego (porażenie, epizody tężyczki). W badaniach biochemicznych stwierdza się hipokaliemię, hipomagnezemię, hipokalciurię i zasadowicę metaboliczną; stężenia reniny i aldosteronu są podwyższone [46]. Wiadomo, że geny kodujące kotransporter sodowo-chlorkowy, których mutacje są odpowiedzialne za wystąpienie zespołu Gitelmana, są zlokalizowane na chromosomie 16, natomiast w przypadku zespołu Gordona nie zidentyfikowano zmian molekularnych dotyczących tego ko-transportera, a wykazano sprzężenia z chromosomem 1; podłoże genetyczne obu tych chorób wydaje się zatem całkowicie odmienne [45].
Zespół Liddle’a jest w pewnym sensie zaburzeniem „odwrotnym” do PAH1. Podłoże genetyczne tego zespołu stanowią mutacje aktywujące genów kodujących podjednostki nabłonkowego kanału sodowego ENaC, podczas gdy mutacje prowadzące do utraty lub upośledzenia funkcji tego samego kanału są przyczyną PAH1 [47]. Warto wspomnieć, że mutacje aktywujące dotyczą genów kodujących łańcuchy β i γ ENaC (zlokalizowanych na chromosomie 16), podczas gdy mutacje odpowiedzialne za PAH1 dotyczą genów kodujących zewnętrzne części łańcucha α (na chromosomie 16) i łańcucha β. W wyniku mutacji aktywujących dochodzi do nadmiernej ekspresji ENaC w błonie cewki dystalnej, co skutkuje nadmiernym wchłanianiem zwrotnym sodu. Klinicznie zespół manifestuje się ciężkim nadciśnieniem tętniczym z niskim stężeniem reniny i aldosteronu oraz hipokaliemią, zasadowicą i hiperkalciurią o zmiennym nasileniu [45]. Zarówno nadciśnienie, jak i zaburzenia biochemiczne nasilają się z wiekiem, we wczesnym dzieciństwie choroba może przebiegać bezobjawowo. W terapii skuteczne są jedynie leki, których mechanizm działania polega na hamowaniu ENaC: amilorid i triamteren; całkowicie nieskuteczne jest stosowanie spironolaktonu [42, 45].
Zespół Barttera to dziedziczona autosomalnie recesywnie tubulopatia związana z utratą soli, zasadowicą metaboliczną i hipokaliemią, z prawidłowym lub obniżonym ciśnieniem tętniczym, z wtórnie podwyższonymi stężeniami reniny i aldosteronu. W okresie ciąży często stwierdzane jest wielowodzie (polihydramnios), a we wczesnym dzieciństwie obserwuje się poliurię i skłonność do odwodnienia. Przyczyną zaburzeń są defekty dotyczące transportu elektrolitów w części cienkiej ramienia wstępującego pętli nefronu. Wyróżniono pięć typów tego zespołu, wykazujących pewne odrębności przebiegu klinicznego, dla których zidentyfikowano odrębne defekty genetyczne. Typ I, II i IV przebiegają z izo- bądź hipostenurią, w typie I i II stwierdza się ponadto zwiększone wydalanie magnezu i wapnia, zaburzeniem charakterystycznym dla typu IV jest głuchota sensomotoryczna. Typ III zespołu ma najłagodniejszy przebieg kliniczny. W większości przypadków zespołu Barttera dziedziczenie jest autosomalne recesywne, natomiast typ V tego zespołu dziedziczy się autosomalnie dominująco, a mutacje prowadzą do nadmiernej aktywacji receptora wapniowo-wrażliwego (CaSR), który ma istotne znaczenie w regulacji wydzielania parathormonu (PTH). U chorych stwierdza się hipokalcemię z hiperkalciurią, którym towarzyszy niskie stężenie PTH [42].
Podsumowanie
Przedstawione zaburzenia to na ogół choroby występujące bardzo rzadko. Niosą one jednak ze sobą poważne ryzyko związane z jednej strony z obecnością zaburzeń gospodarki elektrolitowej pod postacią zespołu utraty soli (ze wszystkimi jego konsekwencjami, łącznie z zagrożeniem życia dziecka), a z drugiej strony – z rozwojem już we wczesnym dzieciństwie nadciśnienia tętniczego. Niektóre z tych zaburzeń mogą mieć podobny obraz kliniczny i biochemiczny pomimo całkowicie odrębnej przyczyny, a tym samym konieczności zupełnie innego postępowania. Często problemem może być odróżnienie defektu stanowiącego pierwotne podłoże choroby od zaburzeń wtórnych, towarzyszących temu defektowi.
Na zakończenie, aby nawiązać do pierwszej części opracowania, dotyczącej zaburzeń gospodarki wodnej (i w ten sposób uświadomić sobie, że rzeczywistość jest jeszcze bardziej skomplikowana), warto zwrócić uwagę na fakt, że zaburzeniom gospodarki wodnej towarzyszą zazwyczaj zmiany stężeń jonów w surowicy, a zaburzeniom elektrolitowym – odwodnienie lub przewodnienie.
Istnieją ponadto pewne bezpośrednie związki pomiędzy regulacją gospodarki wodnej i elektrolitowej, jak np. wspomniana w początkowej części
opracowania możliwość regulacji funkcji kanałów ENaC przez ADH [3, 4] czy zwiększona sekrecja ADH u pacjentów z niewydolnością kory nadnerczy [48]. W roku 2010 przedstawiono też opis przypadków nabytej moczówki nerkopochodnej w przebiegu wrodzonych chorób nerek, m. in. zespołu Barttera i AME [49].
Założeniem naszej pracy było przedstawienie możliwie szerokiego zakresu zaburzeń gospodarki wodnej (w części I) i sodowej (w części II) spotykanych u pacjentów w wieku rozwojowym, przede wszystkim w aspekcie klinicznym. Stąd też świadomie ograniczono szczegółowe opisy dotyczące podłoża genetycznego i molekularnego omawianych jednostek chorobowych. Osoby zainteresowane tą tematyką zachęcamy do zapoznania się z cytowanymi pozycjami piśmiennictwa, zwłaszcza publikacjami w języku polskim.
Piśmiennictwo
1. Stockand J.D.; New ideas about aldosterone signaling in epithelia; Am. J. Physiol. Renal. Physiol. 2002:282, 559-576
2. Muto S.; Action of aldosterone on renal collecting tubule cells; Curr. Opin. Nephrol. Hypertens. 1995:4, 31-40
3. Snyder P.M.; The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension; Endocr. Rev. 2002:23, 258-275
4. Stockand J.D.; Vasopressin regulation of renal sodium excretion; Kidney Int. 2010:78, 849-856
5. Sudoh T., Kangawa K., Minamino N. et al.; A new natriuretic peptide in porcine brain; Nature 1988:332, 78-81
6. Yasue H., Yoshimura M., Sumida H. et al.; Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure; Circulation 1994:90, 195-203
7. Bogdan M., Sielski S., Grześk G. et al.; Mózgowy peptyd natriuretyczny w niewydolności serca; Forum Kardiologów 2004:9, 1-7
8. Sun Y., Ahokas R.A., Bhattacharya S.K. et al.; Oxidative stress in aldosteronism; Cardiovasc. Res. 2006:71, 300-309
9. Zia A.A., Kamalov G., Newman K.P. et al.; From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation; Hypertens, Res. 2010:33, 1091-1101
10. Bhattacharya S.K., Gandhi M.S., Kamalov G. et al.; Myocardial remodeling in low-renin hypertension: molecular pathways to cellular injury in relative aldosteronism; Curr. Hypertens. Rep. 2009:11, 412-420
11. Whaley-Connell A., Johnson M.S., Sowers J.R.; Aldosterone: role in the cardiometabolic syndrome and resistant hypertension; Prog. Cardiovasc. Dis. 2010:52, 401-419
12. Romer T.E.; Wrodzony przerost nadnerczy. [w:] Endokrynologia w codziennej praktyce lekarskiej; Red. Syrenicz A. Wyd. PAM w Szczecinie 2009, 519-556
13. Lifton R.P., Dluhy R.G., Powers M. et al.; Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase; Nat. Genet. 1992:2, 66-7
14. Mulatero P., Morra Di Cella S., Williams T.A. et al.; Glucocorticoid remediable aldosteronism: low morbidity and mortality in a four-generation Italian pedigree; J. Clin. Endocrinol. Metab. 2002:87, 3187-3191
15. Fallo F., Pilon C., Williams T.A. et al.; Coexistence of different phenotypes in a family with glucocorticoid-remediable aldosteronism; J. Hum. Hypertens. 2004:18, 47-51
16. Stowasser M., Gordon R.D.; Primary aldosteronism: from genesis to genetics; Trends Endocrinol. Metab. 2003:14, 310-317
17. Geller D.S., Zhang J., Wisgerhof M.V. et al.; A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism; J. Clin. Endocrinol. Metab. 2008:93, 3117-3123
18. Mulatero P., Williams T.A., Monticone S. et al.; Veglio F. Is familial hyperaldosteronism underdiagnosed in hypertensive children?; Hypertension 2011:57, 1053-1055
19. Aglony M., Martínez-Aguayo A., Carvajal C.A. et al.; Frequency of familial hyperaldosteronism type 1 in a hypertensive pediatric population: clinical and biochemical presentation; Hypertension 2011:57, 1117-1121
20. Monder C.; Corticosteroids, receptors, and the organ-specific functions of 11 beta-hydroxysteroid dehydrogenase; FASEB J. 1991:5, 3047-3054
21. New M.I., Stoner E., DiMartino-Nardi J.; Apparent mineralocorticoid excess causing hypertension and hypokalemia in children; Clin. Exp. Hypertens. A. 1986:8, 751-772
22. Mantero F., Palermo M., Petrelli M.D. et al.; Apparent mineralocorticoid excess: type I and type II; Steroids 1996:61, 193-196
23. Dave-Sharma S., Wilson R.C., Harbison M.D. et al.; (1998) Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess; J. Clin. Endocrinol. Metab. 1998:83, 2244-2254
24. Nunez B.S., Rogerson F.M., Mune T. et al.; Mutants of 11beta-hydroxysteroid dehydrogenase (11-HSD2) with partial activity: improved correlations between genotype and biochemical phenotype in apparent mineralocorticoid excess; Hypertension 1999:34, 638-642
25. Mew M.I., Dluhy R.G. (tłum. Godlewska P); Nadciśnienie tętnicze wywołane zaburzeniami enzymatycznymi syntezy hormonów steroidowych. [w:] Nadciśnienie hormonalne. Red. Januszewicz W., Sznajderman M., Januszewicz A.; Wyd. Naukowe PWN Warszawa 1997, 205-247
26. Romer T.E.; Postępy w rozpoznawaniu i leczeniu wrodzonego przerostu nadnerczy; Endokrynol. Pol. 2003:54, 631-656
27. Szymczak J., Bohdanowicz-Pawlak A.; Wrodzony przerost nadnerczy wywołany niedoborem 11b-hydroksylazy. Opis przypadku; Endokrynol. Polska 2008:59, 521-528
28. Smyczyńska J., Stawerska R., Hilczer M.; Manifestacja kliniczna różnych postaci wrodzonego przerostu nadnerczy; Klinika Pediatryczna 2011:19, 28-34
29. Geller D.S., Farhi A., Pinkerton N. et al.; Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy; Science 2000:289, 119-123
30. Huyet J., Pinon G.M., Fay M.R. et al.; Structural basis of spirolactone recognition by the mineralocorticoid receptor; Mol. Pharmacol. 2007:72, 563-571
31. Lange M., Maciejewska J., Rumińska M. et al.; Wylew do nadnerczy u noworodków: obraz kliniczny i ultrasonograficzny; Endokrynologia Pediatryczna 2006:5 (Supl. 5), 74 (abstr.)
32. Zgorzalewicz M., Suda P., Toczko A.; Adrenoleukodystrofia u 13-letniego chłopca; Przegląd Pediatryczny 2003:33, 317-320
33. Fichna M., Fichna P., Korman E.; Adrenoleukodystrofia – patogeneza, diagnostyka i leczenie; Endokrynologia Pediatryczna 2004:1(6), 57-67
34. Fichna P., Fichna M., Korman E.; Endokrynologiczna charakterystyka adrenoleukodystrofii u dzieci i młodzieży; Endokrynologia Pediatryczna 2004:1(6), 69-81
35. Aron D.C., Findling J.W., Tyrrel J.B.; Glucocorticoids & adrenal androgens. [w:] Greenspan’s Basic & clinical endocrinology (eighth edition). Red. Gardner D.G., Shoback D.; McGraw Hill Medical 2007, 346-395
36. Furgeson S.B., Linas S.; Mechanisms of type I and type II pseudohypoaldosteronism; J. Am. Soc. Nephrol. 2010:21, 1842-1845
37. Szalecki M., Wójcik E., Domagała Z., Małunowicz E.; Pseudohipoaldosteronizm u niemowląt jako przyczyna zespołu utraty soli; Endokrynologia, Diabetologia i Choroby Przemiany Materii Wieku Rozwojowego 2007:13, 1, 33-36
38. Małunowicz E.M., Szalecki M.; Przejściowy pseudohypoaldosteronizm jako przyczyna utraty soli u 8 niemowląt płci męskiej; Endokrynologia Pediatryczna 2006:5 (Supl. 5), 25 (abstr.)
39. Cheek D.B., Perry J.W.; A salt wasting syndrome in infancy; Arch. Intern. Med. 1958:33, 252-256
40. Manikam L., Cornes M.P., Kalra D., Ford C., Gama R.; Transient pseudohypoaldosteronism masquerading as congenital adrenal hyperplasia; Ann. Clin. Biochem. 2011:48, 380-382
41. Edelheit O., Hanukoglu I., Gizewska M., Kandemir N., Tenenbaum-Rakover Y. et al.; Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism; Clin. Endocrinol. (Oxf). 2005 May:62(5), 547-553
42. Rolim A.L., Lindsey S.C., Kunii I.S. et al.; Ion channelopathies in endocrinology: recent genetic findings and pathophysiological insights; Arq. Bras. Endocrinol. Metabol. 2010:4, 673-681
43. Paver W.K., Pauline G.J.; Hypertension and hyperpotassemia without renal disease in a young male; Med. J. Aust. 1964:2, 305-306
44. Gordon R.D., Geddes R.A., Pawsey C.G. et al.; Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction; Australas Ann. Med. 1970:19, 287-294
45. Litwin M.; Genetyczne aspekty nadciśnienia tętniczego u dzieci. [w:] Wybrane zagadnienia z nefrologii dziecięcej. Red. Grenda R.; Wyd. Lekarskie PZWL Warszawa 2003, 133-148
46. Graziani G., Fedeli C., Moroni L. et al.; Gitelman syndrome: pathophysiological and clinical aspects; QJM 2010:103, 741-748
47. Schild L.; The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism; Nephrologie. 1996:17, 395-400
48. Kamoi K., Tamura T., Tanaka K. et al.; Hyponatremia and osmoregulation of thirst and vasopressin secretion in patients with adrenal insufficiency; J. Clin. Endocrinol. Metab. 1993:77, 1584-1588
49. Bockenhauer D., van’t Hoff W., Dattani M. et al.; Secondary nephrogenic diabetes insipidus as a complication of inherited renal diseases; Nephron Physiol. 2010:116, 23-29








