Endokrynol. Ped. 12/2013;3(44):47-54
DOI: 10.18544/EP-01.12.03.1457
Przejściowe zaburzenia gospodarki węglowodanowej w czasie intensywnego leczenia ostrej białaczki limfoblastycznej u dzieci
1Klinika Endokrynologii i Diabetologii Dziecięcej, Uniwersytet Medyczny w Lublinie
2Klinika Hematologii, Onkologii i Transplantologii Dziecięcej, Uniwersytet Medyczny w Lublinie
Słowa kluczowe: cukrzyca, ostra białaczka limfoblastyczna
Streszczenie
Ostra białaczka limfoblastyczna (acute lymphoblastic leukemia, ALL) jest najczęstszą chorobą rozrostową wieku dziecięcego. Powstaje na skutek klonalnej proliferacji prekursorowych limfocytów B lub T. Ważnymi lekami w leczeniu tej limfoproliferacyjnej choroby są L-asparaginaza oraz steroidy. Oba te leki mogą powodować zaburzenia gospodarki węglowodanowej. Celem pracy jest analiza częstości występowania nieprawidłowości w gospodarce węglowodanowej u dzieci leczonych z powodu ALL. Pacjenci i metody. Analizie poddano dokumentację medyczną 97 pacjentów. Dyskusja i wnioski. Zastosowanie L-asparaginazy oraz steroidów w leczeniu pacjentów z ostrą białaczką limfoblastyczną jest czynnikiem predysponującym do wystąpienia zaburzeń gospodarki węglowodanowej. U 11 pacjentów (11,4%) z wartościami glikemii poniżej 230 mg/dl normalizację glikemii uzyskiwano poprzez zastosowanie diety cukrzycowej. U 9 pacjentów (9,2%) z wartościami glikemii przekraczającymi 250 mg/dl leczeniem z wyboru była dieta oraz insulinoterapia. Ukończenie steroidoterapii wiązało się ze stopniową redukcją dawek insuliny, a następnie zakończeniem insulinoterapii. U żadnego z pacjentów po zakończeniu leczenia przeciwnowotworowego nie obserwowano trwałej cukrzycy
Wstęp
Ostra białaczka limfoblastyczna (acute lymphoblastic leukemia, ALL) jest najczęstszą chorobą rozrostową wieku dziecięcego. Powstaje na skutek klonalnej proliferacji prekursorowych limfocytów B lub T. Częstość występowania ALL wynosi około 4 nowych przypadków białaczki na 100 000 dzieci w wieku do 14 roku życia [1].
Leczenie ALL składa się z czterech etapów i trwa łącznie ok. 3 lat. Protokół leczenia chorych z ALL zależy od kwalifikacji do grup zaawansowania na podstawie objawów klinicznych, wyników badań diagnostycznych i reakcji na wstępne leczenie. W przypadku ALL u dzieci od roku 2002 w Europie stosowany jest protokół ALL-IC (trzy grupy ryzyka: SR – standard risk, ryzyko standardowe, IR – intermediate risk, ryzyko pośrednie, HR – high risk, wysokie ryzyko). Obecnie pacjenci z ostrą białaczką limfoblastyczną leczeni są według Programu ALLIC 2002, który składa się z trzech protokołów leczniczych: I, Mm i II [1–4].
I etap terapii to leczenie indukujące (indukcja remisji), mające na celu osiągnięcie prawidłowego stanu dziecka, prawidłowego mielogramu oraz obrazu krwi obwodowej i płynu mózgowo-rdzeniowego. Na tym etapie pełną remisję choroby uzyskuje się u 97–98% dzieci. Stosowane są: winkrystyna, antracykliny (dauno- lub doksorubicyna), L-asparaginaza, glikokortykosterydy (prednizon, deksametazon) oraz leczenie wspomagające, które obejmuje profilaktykę zakażeń, nefropatii moczanowej i leukostazy (w białaczkach hiperleukocytarnych) oraz podawanie koncentratu krwinek czerwonych przy niedokrwistości.
II etap to leczenie konsolidujące (konsolidacja remisji), trwające kilka kolejnych miesięcy, w celu redukcji resztkowych komórek białaczkowych: cyklofosfamid, arabinozyd cytozyny, metotreksat podawany w dużych dawkach, 6-merkaptopuryna, 6-tioguanina. Ten etap kończy się tzw. reindukcją (powtórzeniem zestawu leków z I etapu).
III etap to profilaktyka lub leczenie zajęcia OUN: duże dawki metotreksatu (do płynu mózgowo-rdzeniowego jako profilaktyka zajęcia ośrodkowego układu nerwowego) w monoterapii lub razem z arabinozydem cytozyny i prednizonem.
IV etap to leczenie podtrzymujące: przez co najmniej 2 lata od rozpoznania ALL: 6-merkaptopuryna doustnie, metotreksat okresowo (najczęściej raz w tygodniu).
Ważnymi lekami w leczeniu ALL są L-asparaginaza oraz steroidy. Oba te leki mogą powodować zaburzenia gospodarki węglowodanowej.
L-asparaginaza jest hydrolazą, enzymem katalizującym rozszczepienie L-asparaginy do kwasu asparaginowego i amoniaku. Eliminacja L-asparaginy z otoczenia komórek nowotworowych prowadzi do zahamowania cyklu komórkowego w fazie G1, zahamowania syntezy DNA i białek, apoptozy komórek i w efekcie do śmierci komórki. Zahamowanie syntezy białek jest przyczyną większości powikłań obserwowanych w trakcie leczenia L-asparaginazą, zarówno preparatem w postaci natywnej, jak i pegylowanej, takich jak upośledzenie funkcji wątroby, trzustki, ośrodkowego układu nerwowego. Zaburzenia zewnątrzwydzielniczej czynności trzustki przebiegają ze wzrostem poziomu amylazy i lipazy w surowicy krwi, zaburzenia czynności wewnątrzwydzielniczej prowadzą do hiperglikemii [5, 6].
Przyczyny hiperglikemii wynikłe ze stosowania L-asparaginazy są złożone. Uważa się , że L-asparaginaza powoduje: 1) zmiany w obrębie receptorów insulinowych: hamuje ich syntezę oraz przyspiesza degradację, prowadzi do modyfikacji struktury receptorów z obniżeniem ich aktywności, 2) zmniejszenie dostępności struktur receptorowych w wyniku ich przemieszczenia do przestrzeni wewnątrzkomórkowej, 3) zmianę struktury cząsteczki insuliny i w efekcie obniżenie jej aktywności, 4) podwyższenie stężenia glukagonu i wzrost uwalniania glukozy z glikogenu, 5) podkreśla się również możliwość indukowania p/c przeciwinsulinowych przez L-asparaginazę [7–10].
Mechanizm przeciwnowotworowego działania glikokortykosteroidów nie został dokładnie poznany, ale wiadomo, że na komórkach limfoidalnych znajdują się receptory dla sterydów, a ich liczba na limfoblastach białaczkowych jest znacznie większa niż na limfoblastach prawidłowych. Do najczęściej spotykanych objawów niepożądanych wynikających z zastosowania steroidoterapii należą cukrzyca i posterydowy zespół Cushinga.
Uważa się, że za rozwój cukrzycy sterydowej odpowiedzialnych jest kilka czynników. Jednym z głównych mechanizmów jest wzrost insulinooporności obwodowej. Steroidy prowadzą do insulinooporności przede wszystkim poprzez upośledzenie fosforylacji reszt tyrozynowych receptora insulinowego, a także zahamowanie syntezy i fosforylacji wewnątrzkomórkowego białka IRS-1 [11, 12]. Glikokortykosteroidy przyczyniają się do wzrostu glikemii również na drodze aktywacji transkrypcji enzymów biorących udział w glukoneogenezie wątrobowej przy równoczesnym zwiększeniu podaży substratów potrzebnych do glukoneogenezy na skutek aktywacji procesów katabolicznych (proteolizy i lipolizy) [13]. Steroidy hamując aktywność syntazy glikogenowej zmniejszają utylizację glukozy) [14]. Istnieją też doniesienia o hamującym wpływie glikokortykoidów na sekrecję insuliny przez komórki beta wysp trzustkowych. Jest to efekt nasilenia rozpadu transporterów glukozy GLUT-2 oraz zwiększenia ekspresji receptora alfa -2-adrenergicznego, co prowadzi do upośledzenia wydzielania insuliny [15]. Połączenie preparatów L-asparaginazy ze steroidami istotnie zwiększa częstość wystąpienia zaburzeń gospodarki węglowodanowej.
Celem pracy jest analiza częstości występowania nieprawidłowości w gospodarce węglowodanowej u dzieci leczonych z powodu ALL.
Pacjenci i metody
Analizie poddano dokumentację medyczną 97 pacjentów leczonych z powodu ostrej białaczki limfoblastycznej w latach 2006–2009 w Klinice Hematologii, Onkologii i Transplantologii Dziecięcej, a konsultowanych w Klinice Endokrynologii i Diabetologii Dziecięcej UM w Lublinie. Grupę badaną stanowiło 57 chłopców i 40 dziewczynek w wieku od 2 do 18 lat, leczonych według Programu ALLIC 2002. U wszystkich w leczeniu zastosowano L-asparaginazę i steroidy. W I protokole leczniczym zastosowano 8 dawek L-asparaginazy (5000j / m2) lub 2 dawki pegylowanej postaci L-asparaginazy 1000j/m2 oraz 38-dniową terapię prednizonem w dawce 40 mg/m2. W II protokole leczniczym były 4 dawki L-asparaginazy lub 1 dawka pegylowanej postaci L-asparaginazy oraz 28-dniowa kuracja dexametasonem w dawce 10 mg/m2 (ryc. 1).
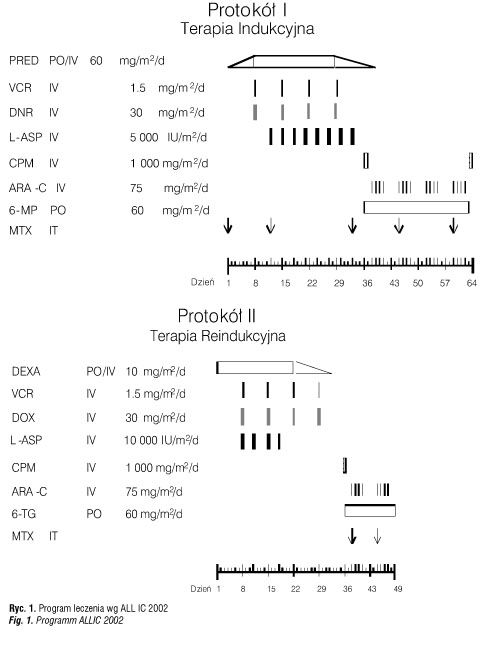
Badania poziomu glikemii wykonywano we krwi żylnej u wszystkich dzieci w czasie leczenia ALL metodą enzymatyczną przy użyciu analizatora COBAS Integra 400. W przypadku rozpoznania cukrzycy według kryteriów IDF glikemię kontrolowano we krwi włośniczkowej przy pomocy glukometrów. U dzieci oznaczano również glukozurię i poziom ciał ketonowych w moczu przy użyciu testów paskowych. Oznaczano też hemoglobinę glikowaną metodą immunoenzymatyczną (Abbott). Aby ocenić zewnątrzwydzielniczą funkcję trzustki, oznaczano poziomy amylazy i lipazy metodą kolorymetryczną przy pomocy analizatora biochemicznego Siemens Advia 1200.
Wyniki
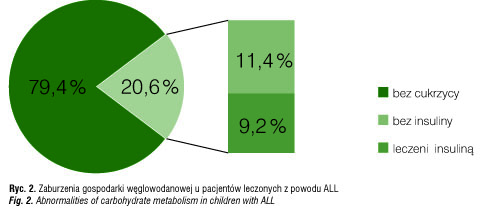
Zaburzenia funkcji wewnątrzwydzielniczej trzustki w postaci cukrzycy (glikemie na czczo ≥126 mg/dl, między posiłkami ≥ 200 mg/dl) stwierdzono u 20 pacjentów, co stanowi 20,6% całej analizowanej grupy. U 18 spośród nich wystąpiły typowe dla cukrzycy objawy w postaci polidypsji i poliurii. U 2 pacjentów nie obserwowano żadnych objawów związanych z hiperglikemią, a podwyższone stężenia glukozy stwierdzono w trakcie wykonywania rutynowych badań laboratoryjnych. U żadnego pacjenta nie wystąpiła dysfunkcja części zewnątrzwydzielniczej trzustki – świadczyły o tym prawidłowe poziomy amylazy i lipazy w surowicy krwi. W grupie 20 pacjentów, u których wystąpiła cukrzyca, 14 było w trakcie protokołu I, a 6 innych w trakcie protokołu II.
U 11 dzieci z glikemiami <230 mg/dl (11,4% całej analizowanej grupy) po zastosowaniu diety cukrzycowej oraz ograniczeniu podaży glukozy we wlewach kroplowych osiągnięto normalizację stężenia glukozy: glikemie na czczo wynosiły od 89 mg/dl do 103 mg/dl, glikemie poposiłkowe: 122 mg/dl–145 mg/dl.
U 9 pacjentów, co stanowiło 9,2% całej analizowanej grupy, stwierdzono: glikemie > 250 mg/dl, typowe dla cukrzycy objawy (polidypsja, poliuria), obecność glukozurii i ketonurii przy prawidłowym pH krwi. Nie obserwowano u nich kwasicy ketonowej. Troje z tych dzieci pochodziło z rodzin, w których występowała cukrzyca typu 2 (ryc. 2).
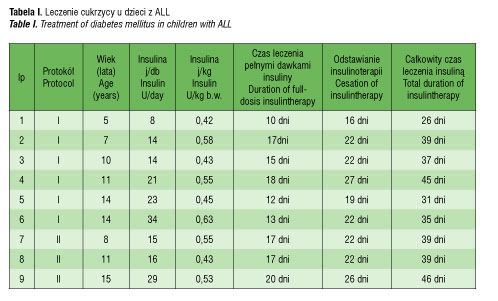
U 5 z tych dzieci stwierdzono cechy dojrzewania: u 3 IV fazę dojrzewania wg skali Tannera, u 2 II fazę. Może mieć to związek ze zwiększonym ryzykiem rozwoju cukrzycy. W trakcie protokołu I było 6 z tych pacjentów, a 3 innych w trakcie protokołu II. Zastosowano u nich oprócz diety cukrzycowej insulinoterapię. W leczeniu stosowano insulinę NPH oraz insulinę krótko działającą (regular) przez okres 10 do 27 dni. Insulinę NPH podawano w dwu dawkach – rano i wieczorem, a insulinę krótko działającą w 3 dawkach – przed głównymi posiłkami. Zapotrzebowanie na insulinę wynosiło 0,42–0,63 j/kg masy ciała/dobę. Średnie zapotrzebowanie na insulinę w całej grupie wynosiło 0,51±0,07 j/kg m.c., w grupie leczonej wg protokołu I – 0,51±0.08 j/kg m.c, w grupie leczonej wg protokołu II – 0,5±0,05 j/kg m.c (tab. 1).
Od momentu rozpoczęcia insulinoterapii uzyskano stopniowo ustąpienie polidypsji i poliurii, ujemne wyniki gluko- i ketotestów w moczu oraz normalizację stężenia glukozy. U wszystkich pacjentów objawy cukrzycy ustąpiły wraz z zakończeniem sterydoterapii. W czasie trzyletniej obserwacji u żadnego dziecka nie obserwowano rozwoju cukrzycy.
Dyskusja
Połączenie preparatów L-asparaginazy ze steroidami, stosowane równocześnie w protokołach leczniczych, istotnie zwiększa częstość wystąpienia zaburzeń gospodarki węglowodanowej. Ryzyko rozwoju cukrzycy związane ze steroidoterapią jest proporcjonalne do wielkości przyjmowanej dawki sterydu. Każdy wzrost dawki prednizolonu o 0,01 mg/kg/db wiąże się z 5% wzrostem ryzyka rozwoju choroby [16–18]. Ryzyko wystąpienia cukrzycy jest większe, gdy pacjent ma nadmierną masę ciała, wcześniej występowała u niego nieprawidłowa glikemia na czczo lub nieprawidłowa tolerancja glukozy oraz w przypadku występowania cukrzycy w rodzinie [16]. W badanej przez nas grupie u 3 pacjentów w wywiadzie stwierdzono występowanie cukrzycy w rodzinie i u każdego z tych dzieci podczas leczenia ALL wystąpiła cukrzyca. U wszystkich dzieci przed rozpoczęciem leczenia poziomy glikemii były prawidłowe. Ważny jest również wiek rozpoczęcia terapii sterydami. W okresie pokwitania i starzenia się organizmu występuje „fizjologiczna” insulinooporność, co sprzyja rozwojowi cukrzycy [19, 20].
U 6 dzieci w badanej przez nas grupie do rozwoju cukrzycy doszło w trakcie protokołu I. W tym schemacie leczenia stosowany jest prednizolon oraz oprócz innych leków daunorubicyna. W protokole II stosowany jest deksametazon. Według niektórych badań stosowanie deksametazonu bardziej predysponuje do rozwoju cukrzycy niż stosowanie prednizonu [21].
Postępowanie lecznicze w cukrzycy sterydowej w odniesieniu do naszych pacjentów zależało od nasilenia hiperglikemii. Przy glikemiach poniżej 230 mg/dl stosowano z zadowalającym efektem dietę cukrzycową, przy wyższych glikemiach (w badanej grupie wartości powyżej 250 mg/dl) stosowano dietę i insulinoterapię. Według Gittoesa i wsp. u osób dorosłych stosujących przewlekle sterydy przy glikemiach do 216 mg/dl możliwe jest leczenie dietą, przy wartościach między 216 a 306 mg/dl leczenie dietą i doustnymi lekami hipoglikemizującymi, natomiast powyżej 306 mg/dl wskazane jest leczenie insuliną oraz dietą. Zdaniem Gittoesa w cukrzycy wynikłej ze stosowania wysokich dawek sterydów (powyżej dawki równoważnej 40 mg prednizolonu/dzień) leczeniem z wyboru jest jednak insulinoterapia [15].
Ze względu na podkreślane przez wielu autorów podobieństwo w mechanizmie powstawania cukrzycy sterydowej i cukrzycy typu 2 u osób dorosłych z nieznacznie podwyższonymi glikemiami celowe wydaje się stosowanie w uzasadnionych przypadkach doustnych leków hipoglikemizujacych [22]. Jednak ze względu na brak dużych randomizowanych badań klinicznych potwierdzających bezpieczeństwo i skuteczność stosowania leków doustnych w cukrzycy sterydowej wielu autorów uważa, że jedynym zalecanym sposobem leczenia jest insulinoterapia [23–30]. W odniesieniu do dzieci insulinoterapia w połączeniu z dietą cukrzycową jest jedyną skuteczną i bezpieczną metodą leczenia. Dawki insuliny, ilość wstrzyknięć należy indywidualizować w zależności od wartości glikemii.
U wszystkich obserwowanych przez nas pacjentów objawy cukrzycy ustąpiły wraz z zakończeniem steroidoterapii. Przemijający charakter cukrzycy sterydowej u dzieci leczonych z powodu ALL obserwowali również inni autorzy [31–33].
Według Mohna i wsp. chemioterapia może uszkadzać część zewnątrzwydzielniczą trzustki [34].
Podobnie jak w naszych badaniach brak zaburzeń części zewnątrzwydzielniczej trzustki podczas leczenia ALL obserwowała Angela M. Spinola-Castro i wsp. [19]. Jednak zdaniem innych autorów podczas stosowania L-asparaginazy u 2–16% dzieci występuje zapalenie trzustki [35, 36].
Wnioski
1. Zastosowanie L-asparaginazy oraz steroidów w leczeniu pacjentów z ostrą białaczką limfoblastyczną jest czynnikiem predysponującym do wystąpienia zaburzeń gospodarki węglowodanowej.
2. U pacjentów z wartościami glikemii poniżej 230 mg/dl normalizację glikemii uzyskiwano poprzez zastosowanie diety cukrzycowej.
3. U pacjentów z wartościami glikemii przekraczającymi 250 mg/dl leczeniem z wyboru była dieta oraz insulinoterapia.
4. Ukończenie steroidoterapii wiązało się ze stopniową redukcją dawek insuliny, a następnie zakończeniem insulinoterapii.
5. U żadnego z pacjentów po zakończeniu leczenia przeciwnowotworowego nie obserwowano trwałej cukrzycy.
Piśmiennictwo
1. Den Boer M.L., van Slegtenhorst M., De Menezes R.X. et al.; A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study; Lancet Oncol. 2010:2, 125
2. Bhatia S., Landier W.; Evaluating survivors of pediatric cancer; Cancer J. 2005:11, 340
3. Hunger S.P., Lu X., Devidas M. et al.; Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group; J. Clin. Oncol. 2012:30, 1663
4. Salzer W.L., Devidas M., Carroll W.L. et al.; Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children’s oncology group; Leukemia 2010:24, 355
5. Gaynon P.S., Angiolillo A.L., Carroll W.L. et al.; Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children’s Oncology Group Report; Leukemia 2010:24, 285
6. Laugel V., Escande B., Entz-Werle N., Mazingue F., Ferster A., Bertrand Y., Missud F., Lutz P.; Severe acute pancreatitis in children receiving asparaginase: Multicenter retrospective study [Pancréatites aiguës sévères à l’asparaginase chez l’enfant: Étude rétrospective multicentrique]; Archives de Pediatrie 2005:12, 34
7. Alves C., Chaves C., Souza M.; Transient diabetes mellitus related to L-asparaginase therapy [Diabetes melito transitório relacionado à terapia com L-asparaginase] a Serviço de Endocrinologia Pediátrica, Faculdade de Medicina, Universidade Federal da Bahia, Salvador; BA Arquivos Brasileiros de Endocrinologia e Metabologia 2007:51, 635
8. Carpentieri U., Balch M.T.; Hyperglycemia associated with the therapeutic use of L-asparaginase: Possible role of insulin receptors; J. Pediatr. 1978:93, 775
9. Rao S.P., Castells S.; Hyperglucagonemia In L-asparaginase induced diabetes mellitus; Am. J. Pediatr. Hematol. Oncol. 1986:8, 83
10. Weetman R.; Latent onset of clinical pancreatitis in children receiving-asparaginase therapy; Cancer 1974:19, 581
11. Meyer G., Badenhoop K.; Glucocorticoid- induced insulin resistance and diabetes mellitus. Receptor-, postreceptor mechanism, local cortisone action, and new aspects of antidiabetic therapy; Med. Klin. 1998:5, 266
12. Nicod N., Giusti V., Besse C. et al.; Metabolic adaptations in dexamethasone-induced insulin resistance in healthy volunteers; Obesity Research 2003:11, 625
13. Yoon J., Puigserver P., Chen G. et al.; Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1; Nature 2001:413, 131
14. Iwamoto T., Kagawa Y., Naito Y. et al.; Steroid-induced diabetes mellitus and related risk factors In patients with neyrologic diseases; Pharmacotherapy 2004:24, 508
15. Gittoes N., Ayuk J., Ferner R.; Drug-induced diabetes. W: Pikkuo J., Wiliams G.N. (red.) Textbook of Diabetes 1; Blackwell Science 2003:26, 8
16. Knoderer H.M., Robarge J., Flockhart D.A.; Predicting asparaginase-associated pancreatitis; Pediatr. Blood Cancer 2007:49, 634
17. Venkatraman R., Jayashree M., Singhi S. et al.; Hyperglycemic hyperosmolar nonketotic syndrome in a child with acute lymphoblastic leukemia undergoing induction chemotherapy: Case report; J. Pediatr. Hematol/Oncol. 2005:27, 234
18. Hsu Y.J., Chen Y.C., Ho C.L. et al.; Diabetic ketoacidosis and persistent hyperglycemia as long-term complications of L-asparaginase-induced pancreatitis; Zhonghua Yi Xue Za Zhi (Taipei) 2002:65, 441
19. Siviero-Miachon A.A., Spinola-Castro A.M., Guerra-Junior G.; Detection of metabolic syndrome features among childhood cancer survivors: A target to prevent disease; Vasc. Health Risk Manag. 2008:4, 825
20. Hoffmeister P.A., Storer B.E., Sanders J.E.; Diabetes mellitus in long-term survivors of pediatric hematopoietic cell transplantation; J. Pediatr. Hematol/Oncol. 2004:26, 81
21. Belgaumi A.F., Al-Bakrah M., Al-Mahr M. et al.; Dexamethasone-associated toxicity during induction chemotherapy for childhood acute lymphoblastic leukemia is augmented by concurrent use of daunomycin; Cancer. 2003:97, 2898
22. Tatoń J.; Doustne leki hipoglikemizujące. W: Tatoń J., Czech A. Diabetologia; PZWL Warszawa 2001:1, 338
23. Roberson J.R., Raju S., Shelso J. et al.; Diabetic ketoacidosis during therapy for pediatric acute lymphoblastic leukemia; Pediatr. Blood Cancer. 2008:50, 1207
24. Howard S.C., Pui C.H.; Endocrine complications in pediatric patients with acute lymphoblastic leukemia; Blood Rev. 2002:16, 225
25. Weiser M.A., Cabanillas M.E., Konopleva M. et al.; Relation between the duration of remission and hyperglycemia during induction chemotherapy for acute lymphocytic leukemia with a hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone/methotrexate-cytarabine regimen; Cancer. 2004:100, 1179
26. Baillargeon J., Langevin A.M., Mullins J. et al.; Transient hyperglycemia in Hispanic children with acute lymphoblastic leukemia; Pediatr. Blood Cancer. 2005:45, 960
27. Cetin M., Yetgin S., Kara A. et al.; Hyperglycemia, ketoacidosis and other complications of L-asparaginase in children with acute lymphoblastic leukemia; J. Med. 1994:25, 219
28. Sonabend R.Y., McKay S.V., Okcu M.F. et al.; Hyperglycemia during induction therapy is associated with increased infectious complications in childhood acute lymphocytic leukemia; Pediatr. Blood Cancer. 2008:51, 387
29. Lowas S.R., Marks D., Malempati S.; Prevalence of transient hyperglycemia during induction chemotherapy for pediatric acute lymphoblastic leukemia; Pediatr. Blood Cancer. 2009:52, 814
30. Neville K.A., Cohn R.J., Steinbeck K.S. et al.; Hyperinsulinemia, impaired glucose tolerance, and diabetes mellitus in survivors of childhood cancer: Prevalence and risk factors; J. Clin. Endocrinol. Metab. 2006:91, 4401
31. Parlman K., Ehrlich R.M.; Steroid diabetes in Childhood; Am. J. Child. 1982:136, 64
32. Skomra S., Przybylska T.; Przejściowa cukrzyca z kwasicą ketonowa w przebiegu stosowania L-asparaginazy u dziecka z ostrą białaczką limfoblastyczną; Pol. Tyg. Lek. 1992:47, 31
33. Wang Y.J., Chu H.Y., Shu S.G. et al.; Hyperglycemia induced by chemotherapeutic agents used in acute lymphoblastic leukemia: Report of three cases; Zhonghua Yi Xue Za Zhi (Taipei) 1993:51, 457
34. Mohn A., DiMarzio A., Capanna R. et al.; Persistence of impaired pancreatic β-cell function in children treated for acute lymphoblastic leukaemia; Lancet. 2004:363, 127
35. Top P.C., Tissing W.J., Kuiper J.W. et al.; L-asparaginase-induced severe necrotizing pancreatitis successfully treated with percutaneous drainage; Pediatr. Blood Cancer. 2005:44, 95
36. Kourti M., Tragiannidis A., Makedou A. et al.; Metabolic syndrome in children and adolescents with acute lymphoblastic leukemia after the completion of chemotherapy; J. Pediatr. Hematol/Oncol. 2005:27, 499








