Endokrynol. Ped. 11/2012;3(40):67-80
DOI: 10.18544/EP-01.11.03.1392
Powikłania endokrynologiczne po allogenicznym przeszczepieniu komórek krwiotwórczych w okresie rozwojowym – prezentacja przypadku i omówienie literatury
1Klinika Endokrynologii Dzieci i Młodzieży Katedry Pediatrii, Polsko-Amerykańskiego Instytutu Pediatrii, Collegium Medicum Uniwersytetu Jagiellońskiego w Krakowie; (w latach 2000-2010) Zakład Transplantologii, Katedry Immunologii Klinicznej i Transplan
2Klinika Endokrynologii Dzieci i Młodzieży Katedry Pediatrii, Polsko-Amerykańskiego Instytutu Pediatrii, Collegium Medicum Uniwersytetu Jagiellońskiego w Krakowie
Słowa kluczowe: allogeniczne przeszczepienie komórek krwiotwórczych, powikłania endokrynologiczne, funkcja i morfologia tarczycy, zaburzenia wzrastania i dojrzewania płciowego, cukrzyca, potransplantacyjna metaboliczna choroba kości
Streszczenie
Powikłania endokrynologiczne po przeszczepieniu hematopoetycznych komórek krwiotwórczych (PKK) są jednymi z częstszych odległych powikłań tego leczenia. Mogą pojawiać się wiele lat po ostatecznym zakończeniu leczenia choroby, która była wskazaniem do PKK i niejednokrotnie wymagają leczenia do końca życia pacjenta. Prezentujemy 17,5-letnią pacjentkę z wieloma powikłaniami endokrynologicznymi po leczeniu ostrej białaczki limfoblastycznej rozpoznanej w wieku 10 lat. Terapię rozpoczęto zgodnie z protokołem HR ALL IC-2000, osiem miesięcy później w całkowitej remisji wykonano PKK od dawcy rodzinnego całkowicie zgodnego w układzie HLA. W przygotowaniu do PKK stosowano frakcjonowane naświetlanie całego ciała i wysokodawkowaną chemioterapię. Po transplantacji wystąpiły liczne wczesne i późne powikłania: infekcje o etiologii wirusowej, bakteryjnej i grzybiczej, zespół wszczepienny z ostrą niewydolnością nerek i niewydolnością oddechową, choroba przeszczep p-gospodarzowi oraz ostre zapalenie trzustki. Równocześnie obserwowano liczne powikłania endokrynologiczne: i) przemijającą hiperglikemię w pierwszych dniach po PKK, a następnie cukrzycę z narastającą insulinoopornością, wymagającą leczenia insuliną od 6 miesiąca po PKK, ii) jawną pierwotną niedoczynność tarczycy leczoną L-tyroksyną od 4 miesiąca po PKK, iii) przemijającą hyponatriemię w przebiegu zespołu SIADH, iv) niedobór hormonu wzrostu, który substytuowany był od 2 roku po PKK do 16 r.ż. zgodnie z decyzją pacjentki z okresem 7-miesięcznej przerwy w leczeniu z uwagi na nefropatię błoniastą w przebiegu cGVHD, v) opóźnione pokwitanie z następowym wtórnym brakiem miesiączki, wymagające zastępczej terapii hormonalnej estrogenowo-progesteronowej, vi) niską gęstość mineralną kości w kilkukrotnych badaniach densytometrycznych pomimo suplementacji wapnia i witaminy D3. Istotne powikłania endokrynologiczne po PKK, które wystąpiły u pacjentki, najpewniej były ubocznym skutkiem radioterapii kondycjonującej oraz działań ubocznych stosowanej farmakoterapii, ze szczególnym uwzględnieniem przewlekłej sterydoterapii, oraz następstwem innych niż endokrynologiczne powikłań potransplantacyjnych
Wstęp
Od lat osiemdziesiątych ubiegłego stulecia przeszczepienie allogenicznych komórek krwiotwórczych (allo PKK) jest ratującą życie procedurą
leczniczą stosowaną u dzieci z rozpoznaniem schorzeń nowotworowych oraz nienowotworowych: hematologicznych, metabolicznych i genetycznych. W ostatnich latach ze względu na poszerzenie wskazań do PKK jak i coraz doskonalsze metody lecznicze znacznie wzrasta liczba pacjentów wyleczonych, szczególnie w grupie osób młodych, mających perspektywę długoletniego przeżycia. Prognozuje się, że do roku 2020 liczba wyleczonych osób po PKK wzrośnie do pół miliona [1]. Dając szansę wyleczenia wielu schorzeń pierwotnie źle rokujących, PKK niesie ryzyko wielorakich, odległych powikłań. Spośród nich należy zwrócić szczególną uwagę na powikłania endokrynologiczne, które stwierdza się u 20–50% osób wyleczonych, a mogą pojawić się wiele lat po ostatecznym zakończeniu leczenia choroby, która była wskazaniem do PKK [2, 3]. Do czynników ryzyka uszkodzenia komórek gruczołów dokrewnych zalicza się działanie wysokodawkowanej radio- i chemioterapii w przygotowaniu do transplantacji, ale również działanie wielu leków stosowanych wcześniej w leczeniu choroby podstawowej oraz podawanych w leczeniu powikłań poprzeszczepowych [4]. Również pierwotnie nieendokrynologiczne powikłania PKK mogą mieć znaczący wpływ na funkcjonowanie gruczołów dokrewnych, które odgrywają ogromną rolę w utrzymaniu homeostazy ustroju. Niemałe znaczenie ma wiek i stan kliniczny osoby kwalifikowanej do PKK. Do najczęściej obserwowanych endokrynologicznych powikłań poprzeszczepowych w grupie dzieci i młodzieży zalicza się zaburzenia funkcjonowania tarczycy, uszkodzenie gonad oraz zaburzenia wzrastania wynikające nie tylko z uszkodzenia komórek przysadki mózgowej czy tarczycy, ale również układu kostnego [3,5]. W publikacjach ostatniej dekady przedstawiany jest również problem zaburzeń metabolizmu węglowodanów i tłuszczy, nadciśnienia oraz przemiany kostnej [6–9].
Celem pracy jest prezentacja klinicznego przypadku 17,5-letniej pacjentki z licznymi powikłaniami endokrynologicznymi po leczeniu onkologicznym obejmującym allogeniczne PKK oraz omówienie tych powikłań w oparciu o najnowszą literaturę.
Przypadek kliniczny
Pacjentka w wieku 10 lat została przyjęta do Kliniki Onkologii i Hematologii Dziecięcej Polsko-Amerykańskiego Instytutu Pediatrii z podejrzeniem białaczki w stanie ogólnym dość dobrym.
W badaniu fizykalnym stwierdzono ogólne osłabienie, bladość powłok skórnych, wybroczyny na skórze, powiększoną wątrobę i śledzionę, utrzymującą się gorączkę. W ocenie auksologicznej: wzrost znajdował się na poziomie 50 centyla i był zgodny z prognozą wzrostu obliczoną na podstawie wzrostu rodziców – MPH (mid parental height), masa ciała była proporcjonalna do wzrostu, rozwój płciowy był przedpokwitaniowy.
Rozpoznano ostrą białaczkę limfoblastyczną – ALL (acute lymphoblastic leukemia) wysokiego ryzyka (L-43600, pro-B2 z koekspresją CD15, t(4;11) i rozpoczęto leczenie wg programu HR IC-2002. Remisję stwierdzono po 6 tygodniach leczenia. W 2 miesiącu leczenia po 6 dawce L-asparaginazy wystąpiło jego powikłanie, tj. ostre zapalenie trzustki, przebiegające bez hiperglikemii. Wywiad rodzinny pacjentki nie był obciążony cukrzycą.
W wieku 10 lat i 9 miesięcy w remisji ALL przeprowadzono procedurę allo PKK od dawcy rodzinnego zgodnego w układzie HLA. Leczenie kondycjonujące obejmowało frakcjonowane naświetlanie całego ciała – TBI (total body irradiation) w całkowitej dawce 12Gy oraz wysokodawkowaną chemioterapię, tj. etoposid w dawce 60 mg/kg (łączna dawka 2340 mg). W profilaktyce choroby przeszczep przeciwko gospodarzowi stosowano Cyklosporynę A (CsA) w dawce początkowej 3 mg/kg/d, którą następnie modyfikowano w oparciu o oznaczenia stężeń CsA w surowicy krwi (oznaczenia 2 x w tygodniu). W dobie 0/+1 procedury przeszczepiono łącznie 3,26 x 106 /kg m.c. biorcy komórek CD34+ pobranych uprzednio ze szpiku i krwi obwodowej dawcy po przeprowadzonej mobilizacji G-CSF (granulocyte-colony stimulating factor) w dawce 2 x 5 μg/kg m.c. Odbudowa krwiotworzenia wystąpiła w dobie +11, kiedy to stwierdzono leukocyty > 1,0 x 109/l i granulocyty > 0,5 x 109/l. Liczbę płytek > 20 x 109/l obserwowano w dniu +13, zaś > 50 x 109/l w dniu +14.
Po transplantacji wystąpiły liczne wczesne powikłania. W dobie +5 wystąpiła gorączka o nieznanej etiologii, którą leczono antybiotykami o szerokim spektrum, lekami p-grzybiczymi i p-wirusowymi oraz preparatami immunoglobulin, a od dnia +7 G-CSF w dawce 5 µg/kg/d. Z uwagi na mucositis pacjentka otrzymywała od -2 doby całkowite żywienie pozajelitowe. Od doby +10 wystąpiły objawy zespołu wszczepiennego: skórna postać ostrej GvHD w stopniu III, niewydolność oddechowa i ostra niewydolność nerek. Stosowano leczenie
methylprednisolonem we wzrastających dawkach od dawki początkowej 2 mg/kg/d oraz furosemid do dawki 8 mg/kg/d. W dobie +12 wystąpił obrzęk płuc, pacjentka wymagała zastosowania zastępczej wentylacji mechanicznej z okresową podażą 100% tlenu, którą prowadzono do +128 doby po PKK.
W dobie +17 ze względu na objawy rozsianego krwawienia dopęcherzykowego podano jednorazowo methylprednisolon w dawce 10 mg/kg/d, a następnie powrócono do dawki 4 mg/kg/d. W dobie +15 stwierdzono hiperglikemię > 11,2 mmol/l i do leczenia włączono insulinę podawaną dożylnie do dnia +19, kiedy to glikemie znormalizowały się i były prawidłowe przez kolejne 6 miesięcy.
Od dnia +20 obserwowano narastające, oporne na leczenie nadciśnienie tętnicze, w którego leczeniu stosowano enarenal, cordafen, propranolol, nepresol, metoprolol, uropidyl. W dobie +26 rozpoznano idiopatyczne zapalenie płuc i do prowadzonego leczenia p-mikrobiologicznego dołączono p-ciała anty-TNF alfa. Stan pacjentki był ciągle ciężki. Obserwowano pogorszenie wydolności mięśnia sercowego. Prawdopodobnie pacjentka prezentowała wówczas objawy zespołu niskiej trójjodotyroniny, jednakże nie badano poziomu hormonów.
Od doby +30 stwierdzano hiponatremię w przebiegu zespołu nieadekwatnej produkcji hormonu andtydiuretycznego – SIADH (syndrome of inappropriate antydiuretic hormone), poziomy Na wahały się przedziale 128–135 mmol/l i miały tendencję do normalizacji przy zmniejszaniu podaży płynów.
W dobie +87 rozpoznano prawdopodobną grzybicę układową. W jej leczeniu zastosowano kaspofunginę i obserwowano stopniową poprawę stanu ogólnego pacjentki. Była ona nadal hospitalizowana i po dobie +100 obserwowano pojawianie się późnych powikłań poprzeszczepowych.
W 4 miesiącu po PKK rozpoznano pierwotną niedoczynność tarczycy przy poziomie TSH wynoszącym 36,6 uIU/ml i poziomie fT4 wynoszącym 6,0 pmol/l [N 10-25 pmol/l]. Rozpoczęto substytucję L-tyroksyną.
Od 4 miesiąca po PKK obserwowano też dyslipidemię. Pierwszy profil lipidów był następujący: poziom cholesterolu całkowitego wynosił 6,51
mmol/l (N 2,69-5,88), triglicerydów 4,7 mmol/l (N 0,6-2,35), HDL cholesterolu 1,28 mmol/l (N 0,74-1,78) i LDL cholesterolu 3,1 mmol/l (N 1,18-3,62). W leczeniu stosowano cholestryraminę i statyny.
W piątym miesiącu po PKK wystąpiły objawy zapalenia trzustki. Prowadzono leczenie żywieniowe sondą dojelitową oraz stosowano blokery pompy protonowej. Proces zapalny znacznie zaostrzył się po 3 tygodniach, kiedy to rozpoznano nagminne zapalenie przyusznic na podstawie typowych objawów i potwierdzonego kontaktu z osobą będącą w okresie poprzedzającym wystąpienie tej choroby. W 6 miesiącu po PKK pacjentka wymagała leczenia chirurgicznego ropnia okołotrzustkowego.
Równocześnie stwierdzano narastającą hiperglikemię i w 6 miesiącu po PKK rozpoznano cukrzycę. Do leczenia włączono insulinę drogą podskórną. Pacjentka początkowo wymagała podaży insuliny krótkodziałającej do posiłków, a następnie leczenia metodą zintensyfikowanej insulinoterapii przy użyciu insuliny krótkodziałającej jak i o przedłużonym działaniu. Obserwowano stopniowe narastanie zapotrzebowania na insulinę do dawki 2 j//kg m.c./dobę. Ze względu na prowadzone w tym czasie leczenie immunosupresyjne nie oznaczano poziomu przeciwciał typowych dla cukrzycy, poziom peptydu C pozostawał w normie. Od trzeciego roku leczenia w terapii zastosowano ciągły podskórny wlew szybko działającego analogu insuliny przy użyciu osobistej pompy insulinowej. Obserwowano stopniowe narastanie insulinooporności i pogorszenie wyników leczenia choroby. Poziomy HbA1c w pierwszych 4,5 latach trwania cukrzycy wynosiły 6,8–7,2%, następnie powyżej 8%.
W wieku 16 lat, po 5 latach trwania cukrzycy, pacjentka była hospitalizowana w Klinice Endokrynologii Dzieci i Młodzieży celem poprawy kontroli metabolicznej cukrzycy oraz pierwszorazowej edukacji zasad samokontroli. Przy użyciu systemu ciągłego podskórnego monitorowania glikemii osiągnięto normoglikemię przy zapotrzebowaniu na insulinę przekraczającym 2 j/kg m.c./d. Mimo ciągłej adaptacji dawek insuliny do spożywanego posiłku i wysiłku fizycznego kontrola metaboliczna choroby nadal nie była dobra, a zapotrzebowanie na insulinę wzrosło do 3 j/kg/d. Zdecydowano o dodaniu do leczenia metfominy, uzyskując znaczący spadek zapotrzebowania na insulinę oraz poprawę wyników leczenia cukrzycy (ryc 1).
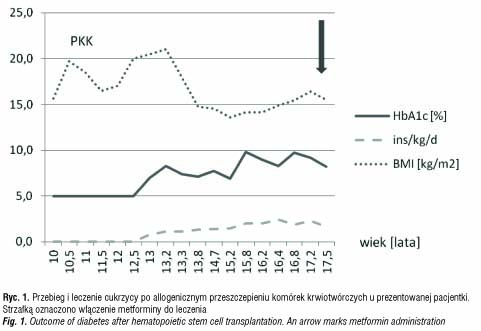
Dwa lata po PKK przeprowadzono diagnostykę w kierunku niedoboru hormonu wzrostu. Parametry auksologiczne w wieku wówczas 12 lat i 9 miesięcy były następujące: wzrost 3,2 SD, wiek wzrostowy 11 lat, wiek kostny 11 lat, masa ciała proporcjonalna do wzrostu (ryc. 2). Tempo wzrastania wynosiło 0 cm/rok i było trudne do obiektywnej oceny z powodu przykurczów stawów kolanowych i biodrowych w przebiegu przewlekłej, skórnej GvHD, leczonej m.in. sterydami. Dojrzewanie oceniono na II stopień wg skali Tannera. Maksymalny wyrzut GH w testach stymulacyjnych wynosił 5,57 ng/ml, zaś w teście nocnego wyrzutu GH 7,2 ng/ml, poziom IGF-1 wynosił 139,3 ng/ml. Badanie MRI okolicy przysadkowo-podwzgórzowej uwidoczniło niejednorodny sygnał przedniego płata przysadki, sugerujący zaburzenia czynnościowe. Rozpoznano somatotropinową niedoczynność przysadki i do leczenia włączono rhGH. Po roku substytucji rhGH zdecydowano się na jej przerwanie ze względu na wystąpienie nefropatii błoniastej w przebiegu przewlekłej GvHD. Po 7-miesięcznej przerwie kontynuowano terapię rhGH do 16 r.ż. uwzględniając odmowną decyzję pacjentki. W trakcie całej terapii uzyskano 9-centymetrowy przyrost wysokości ciała, z pewnym zaburzeniem proporcji ciała będącym skutkiem ubocznym wieloletniej sterydoterapii w leczeniu ostrej i przewlekłej GvHD.
Obserwowano opóźnienie procesu dojrzewania płciowego. Od 11–15 r.ż. stacjonarnie stwierdzano II stopień dojrzewania wg Tannera (Telarche – II), w 15 r.ż. wystąpił spontaniczny progres do stopnia III/IV. Menarche wystąpiło w wieku 15 lat i 4 miesięcy. Następnie pojawiło się 7 regularnych cykli i wtórny brak miesiączki. Obserwowano zahamowanie dojrzewania płciowego w stopniu III/IV wg Tannera. Wykonane w wieku 16 lat i 2 miesięcy badania hormonalne potwierdziły hipogonadyzm hipergonadotropowy (LH 136,5 mIU/ml, FSH 197,89 mIU/ml, estrogeny 21,7 pg/ml). Po podjęciu decyzji o zastępczej terapii hormonalnej estrogenowo-progesteronowej jeszcze przed jej włączeniem wystąpiły dwa nieregularne cykle i ponownie wtórny brak miesiączki. Hormonalną terapię zastępczą (HTZ) rozpoczęto w wieku lat 17 po uzyskaniu zgody pacjentki.
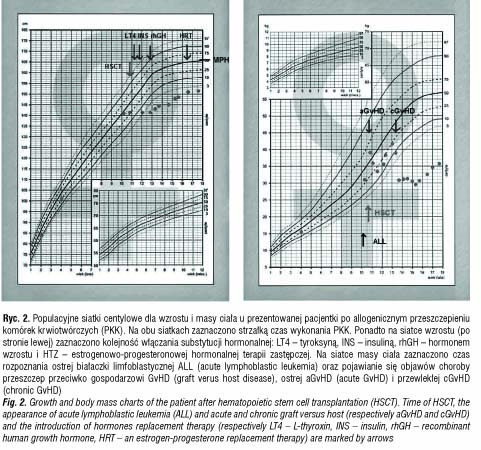
Rok po włączeniu rhGH wykonano pierwsze badanie gęstości mineralnej kości (BMD – bone mineral density), stwierdzając jej znaczne obniżenie. Mimo stosowania preparatów wapnia i witaminy D3 wartości BMD były niskie: total body Z-score zmniejszał się stopniowo z wartości od -2,8 do -4. Poprawę do wartości -3,5 zaobserwowano dopiero w 6 miesięcy po włączeniu HTZ.
Obecnie pacjentka jest w stanie ogólnym dobrym. Uczęszcza do szkoły średniej, podejmuje umiarkowany wysiłek fizyczny. Pozostaje w stałej opiece Poradni Endokrynologicznej, Diabetologicznej, Ginekologicznej, Kardiologicznej, Nefrologicznej i Transplantacyjnej. Otrzymuje na stałe preparaty enzymów trzustkowych do posiłków, insulinę przy użyciu osobistej pompy insulinowej, metforminę, tyroksynę, estrogenowo-progesteronową hormonalną terapię zastępczą drogą przezskórną, digoksynę i inhibitor ACE, ma substytuowany wapń i witaminę D3. Pacjentka pozostaje w euthyreozie, w obrazie USG tarczycy stwierdza się liczne zwłóknienia bez zmian ogniskowych. Ma prawidłowe wartości ciśnienia tętniczego krwi, chociaż w badaniu okulistycznym stwierdza się zmiany na dnie oczu typowe dla nadciśnienia oraz zaćmę podtorebkową, nie stwierdza się zaś mikroangiopatii cukrzycowej. Prawidłowy też jest profil lipidowy pacjentki, mimo odstawienia terapii farmakologicznej.
Dyskusja
Prezentowana pacjentka rozwinęła wszystkie możliwe powikłania endokrynologiczne leczenia onkologicznego ostrej białaczki limfoblastycznej rozpoznanej w dzieciństwie, obejmującego allogeniczne przeszczepienie komórek krwiotwórczych. Zaburzenia endokrynologiczne stwierdzane u dzieci i młodzieży po PKK mają bardzo duże znaczenie dla ich dalszego rozwoju i optymalnej jakości życia w dorosłości. Dlatego też wiedza o ich prewencji i leczeniu jak również odpowiednio przeprowadzanym monitorowaniu w celu ich wykrycia jest również istotna dla wszystkich lekarzy, którzy potencjalnie mogą się opiekować tymi pacjentami, a więc nie tylko dla endokrynologów.
Zaburzenia funkcji i morfologii gruczołu tarczowego
Jednym z najlepiej poznanych powikłań endokrynologicznych po PKK są zaburzenia funkcjonowania i struktury gruczołu tarczowego. W piśmiennictwie dane dotyczące częstości występowania zaburzeń gruczołu tarczowego wahają się od 20 do 58%, a różnice te wynikają z różnej długości czasu trwania obserwacji i różnych liczebności badanych pacjentów [1, 4, 10]. Najczęściej stwierdza się niedoczynność tarczycy, która może wystąpić jako subkliniczna niedoczynność tarczycy, jawna pierwotna niedoczynność tarczycy i wtórna niedoczynność tarczycy wynikająca z wielohormonalnej niedoczynności przysadki. W pierwszym roku po PKK subkliniczna niedoczynność tarczycy występuje u 7–15,5% pacjentów, w większości przypadków jest ona łagodna, dobrze skompensowana i ma tendencję do spontanicznego ustąpienia [5]. Według literatury 11–50% pacjentów po allo PKK rozwija niewydolność tarczycy w bardzo różnym czasie po transplantacji z medianą 48–50 miesięcy [5, 11]. Opierajac się na ostatnim doniesieniu Sanders i jej wsp. z roku2009, obejmującym 30-letni okres obserwacji i leczenia pacjentów po PKK, w około 30% z nich rozwija się różnego typu niedoczynność tarczycy w różnym czasie po transplantacji. 20% z nich wymaga leczenia tyroksyną. U pozostałych 10% obserwowano nadczynność tarczycy oraz zapalenia tarczycy przebiegające z eutyreozą. Niezależnymi czynnikami ryzyka rozwoju schorzeń tarczycy po PKK są wiek pacjentów poniżej 10 lat oraz stosowanie w leczeniu kondycjonującym TBI i wysokich dawek busulfanu. Obie te metody terapii kondycjonującej są obarczone tym samym ryzykiem wystąpienia dysfunkcji gruczołu tarczowego [10].
Zalecane jest badanie poziomu hormonów tarczycy po 6 miesiącach, w późniejszym okresie rokrocznie. Na podstawie wspólnego raportu EBMT/CIBMTR/ASMT średni czas wystąpienia niedoczynności tarczycy to 4 lata po allo PKK, po 20 latach po allo PKK obserwuje się częstszy rozwój raka brodawkowatego tarczycy w tej grupie pacjentów [11].
W przypadku jawnej pierwotnej niedoczynności tarczycy leczeniem z wyboru jest podaż tyroksyny w dawce uzależnionej od poziomu TSH, który zaleca się oznaczać po włączeniu leczenia co 6–8 tygodni, a następnie przy ustaleniu dawki co 6 miesięcy. Według zaleceń poziom TSH powinien w trakcie terapii utrzymywać się blisko dolnych norm z uwagi na większe ryzyko rozwoju raka tarczycy niż w populacji ogólnej. Z tego powodu też zaleca się leczenie subklinicznej niedoczynności pacjentów po PKK, chociaż pod tym względem zdania autorytetów są podzielone [1, 5, 12]. Liczne badania kliniczne nie potwierdziły skuteczności stosowania L-T4 bądź T3 w leczeniu zespołu niskiej trójjodotyroniny, dlatego też nie zaleca się w przypadku jego stwierdzenia stosowania preparatów hormonów tarczycy, chociaż sprawa ta jest ciągle dyskusyjna [3].
Zaburzenia metabolizmu węglowodanów i tłuszczy
Według różnych doniesień u pacjentów po przebytym w dzieciństwie PKK, ze szczególnym uwzględnieniem tych po TBI, stwierdzano insulinooporność aż w 52–83% oraz dyslipidemię w 28–61% [4, 13, 14]. Ryzyko rozwoju cukrzycy u chorych po allogenicznym PKK jest około 3,5 razy większe niż u ich zdrowych krewnych i trzykrotnie wyższe niż w ogólnej populacji Stanów Zjednoczonych [13, 15]. U 8% pacjentów po transplantacji szpiku rozwija się cukrzyca, głównie typu 2.
Jest ona w 40% związana z otyłością (BMI > 30 kg/m2), co stanowi znacznie mniejszy procent niż w ogólnej populacji, natomiast bardzo często jest ona elementem zespołu metabolicznego i wiąże się z nieprawidłowym rozmieszczeniem tkanki tłuszczowej, tj. otyłością brzuszną wynikającą między innymi z niedoboru GH. Ponieważ TBI jest niezależnym czynnikiem ryzyka rozwoju cukrzycy po transplantacji, być może dochodzi do popromiennego uszkodzenia komórek beta wysp trzustkowych, podobnie jak innych komórek gruczołów dokrewnych. U pacjentów po przeszczepie szpiku, u których rozpoznaje się cukrzycę typu 1, prawie nigdy nie stwierdza się typowych autoprzeciwciał [16]. Według danych amerykańskich cukrzycę typu 1 stwierdza się u 0,52% chorych po przebytym PKK, tj. 3-krotnie częściej niż w ogólnej populacji [15].
Z powodu nietypowego jej przebiegu i wieloczynnikowej etiologii trudno jest zakwalifikować cukrzycę rozpoznawaną po leczeniu PKK do określonego typu [17, 18], dlatego też nie ma ustalonego standardu jej leczenia. W literaturze przedstawiane są różne metody leczenia cukrzycy po PKK: wyłącznie dietą, insuliną, jak i doustnymi środkami hipoglikemizującymi [19, 20]. W proponowanych standardach monitorowania pacjentów po PKK w kierunku powikłań odległych nie ma wskazań do rutynowego badania glikemii czy też oznaczania HbA1c.
Na podstawie przytaczanego piśmiennictwa jednakże wydawałoby się to wskazane przynajmniej raz w roku, szczególnie u osób na przewlekłej sterydoterapii z powodu GvHD lub przy wystąpieniu jakichkolwiek objawów sygnalizujących zaburzenia gospodarki węglowodanowej. Monitorowaniem tym powinni być objęci zwłaszcza pacjenci leczeni przewlekle sterydami z powodu GvHD, chociaż rola GvHD jako niezależnego czynnika ryzyka po PKK nie jest jednoznacznie udowodniona [13, 21].
U pacjentów pediatrycznych po PKK występuje większe ryzyko rozwoju zespołu metabolicznego [4, 22, 23]. Wśród czynników ryzyka nadciśnienia występującego u 15–20% spośród wyleczonych po PKK również wymienia się cukrzycę i otyłość, chociaż kluczowymi niezależnymi czynnikami ryzyka jego rozwoju są uszkodzenie nerek, TBI i przebyta infekcja HCV [24, 25]. Dyslipidemię obserwuje się u 9–39% pacjentów po PKK według różnych doniesień [22, 25]. Potwierdzonych czynnikiem ryzyka rozwoju dyslipidemii jest stosowanie TBI w przygotowaniu do transplantacji [26]. Z uwagi na istotnie wyższe ryzyko chorób sercowo-naczyniowych w grupie pacjentów po przebytym PKK wskazane jest szczegółowe monitorowanie każdej ze składowych zespołu metabolicznego oraz leczenie zgodnie z obowiązującymi zasadami leczenia nadciśnienia i dyslipidemii u indywidualnych pacjentów.
Zaburzenia wzrastania
Około 20% pacjentów po przebytym w dzieciństwie lub w okresie dojrzewania PKK osiąga końcowy wzrost poniżej -2 SD, tj. odchylenia standardowego [27]. Ryzyko zaburzeń wzrastania jest szczególnie duże u dzieci małych oraz u tych, które przebyły TBI i napromienianie CSN [27, 28]. Lepszą prognozę wzrastania po PKK mają dziewczynki w porównaniu z chłopcami, zaś pacjenci przygotowywani do PKK tylko wysokodawkowaną chemioterapią osiągają wzrost końcowy porównywalny z prognozą wzrostu opartą na pomiarach wzrostu ich rodziców [27]. Częstość występowania niedoboru hormonu wzrostu po PKK jest różna zdaniem autorów i wynosi od 20 aż do 85% w zależności od typu i czasu przeprowadzanych testów diagnostycznych, ale też w zależności od leczenia poprzedzającego jak i przygotowującego do transplantacji. Wykazano iż stosowanie TBI w przygotowaniu do przeszczepienia w pojedynczej dawce wywiera większy ujemny skutek na osiągany wzrost końcowy niż frakcjonowane napromienianie całego ciała.
Napromienianie powoduje nie tylko uszkodzenie komórek somatotropowych przysadki, ale również uszkodzenie stref wzrostu kości, a w przypadku TBI dotyczy to głównie kręgosłupa. Diagnostyka tego powikłania nie jest ogólnie dostępna i łatwa. Wykazano, iż pacjenci po przebytym w dzieciństwie TBI osiągają niższy wzrost końcowy, ale również mają, i to w większym zakresie niż ich rówieśnicy po PKK bez TBI, niższą wysokość siedzeniową [29]. Dawka stosowana w trakcie TBI nie ma większego wpływu na osiągany wzrost końcowy pacjentów. Nie różni się on bowiem u tych, u których stosowano standardowo 12 Gy w porównaniu do dawek wyższych, tj. 13,75 Gy czy nawet 15 Gy [29]. Do innych przyczyn hormonalnych wystąpienia niskiego wzrostu końcowego może należeć niedobór hormonów tarczycy i hormonów płciowych związanych z występowaniem uszkodzenia gonad w tej grupie pacjentów. Niemałe znaczenie ma stosowanie sterydoterapii w leczeniu niektórych schorzeń onkologicznych, a przede wszystkim długotrwała ich podaż w leczeniu choroby GVHD. U pacjentów, którzy w dzieciństwie przebyli PKK, można wymienić także inne niż hormonalne powody zaburzenia wzrastania. Na pierwszym miejscu są to zaburzenia odżywienia, często towarzyszące terapii onkologicznej poprzedzającej transplantację, występujące też u wszystkich pacjentów we wczesnym okresie PKK, ale również i u tych z powikłaniami poprzeszczepowymi np. przewlekłą, wątrobową, czy jelitową postacią GvHD. W tych przypadkach stwierdza się zaburzenia wątrobowej produkcji IGF-1 [12]. Kolejną dodatkową, rzadko wspominaną, przyczyną zaburzeń wzrastania w tej grupie pacjentów jest deprywacja psychospołeczna wynikająca nie tylko ze świadomości realnego zagrożenia życia, ale również długotrwałej izolacji i unieruchomienia oraz braku możliwości ekspozycji na słońce.
Pacjenci po PKK, u których zdiagnozowano niedobór GH, mają potencjalne wskazania do terapii rhGH. Jednakże nawet z grupy pacjentów, gdzie niedobór GH sięgał 85%, tylko połowa leczona była rhGH [27, 30, 31]. W badaniu Sanders, do którego włączono 90 osób po PKK, wzrost końcowy leczonych rhGH był porównywalny do wzrostu pacjentów ze stwierdzonym niedoborem GH, którzy nie byli leczeni. Najlepsze wyniki osiągnięto o dzieci leczonych przed 10 r.ż. [28]. Do dziś nie są jednoznacznie zdefiniowane działania uboczne terapii rhGH u pacjentów po leczeniu onkologicznym. Na podstawie raportu z 2002 r. u 361 z nich nie stwierdzono ryzyka nawrotu choroby podstawowej, jednakże u 15 osób z tej grupy badanych, najczęściej leczonych uprzednio z powodu białaczki i którym podawano rhGH, wystąpiło podwyższone ryzyko rozwoju nowotworów wtórnych [32].
W cytowanym badaniu jednakże nie u wszystkich leczonych był stwierdzony niedobór GH. Pacjenci po PKK są narażeni na rozwój drugich (wtórnych) nowotworów podobnych do tych, które występują u pacjentów po leczeniu onkologicznym w dzieciństwie. Wyniki badania Sanders z 2005 r., które objęły pacjentów z niedoborem GH po PKK leczonych i nieleczonych rhGH, nie wskazują jednak na wyższe ryzyko rozwoju nowotworów drugich/następnych w grupie leczonych [28]. Mając na uwadze powyższe doniesienia, należy nadal bardzo uważnie kwalifikować pacjentów po PKK do leczenia hormonem wzrostu, rozważając indywidualne potencjalne jego korzyści.
U dzieci po przebytym leczeniu PKK zaleca się ocenę wzrostu masy i ciała przy każdej wizycie kontrolnej i dokładną analizę odpowiednich dla populacji siatek wzrostowych przynajmniej co pół roku po transplantacji. W razie stwierdzenia nieprawidłowości zaleca się podjęcie szczegółowej diagnostyki różnicowej, w tym hormonalnej.
Hipogonadyzm
Hipogonadyzm jest najczęstszym endokrynologicznym późnym powikłaniem po allo PKK. Występuje u niemal wszystkich kobiet, które w wieku dojrzałym przebyły transplantację poprzedzoną wysokodawkowaną radioterapią i chemioterapią z użyciem busulfanu. Jednakże około 40% dziewcząt, które przebyły PKK w okresie prepubertalnym, może mieć zachowaną czynność gonad z możliwością spontanicznie przebiegającego dojrzewania płciowego i wystąpieniem pierwszej miesiączki, obserwuje się u nich jednak przedwczesne wygasanie funkcji jajników [11].
Udowodniono iż dawka promieniowania już poniżej 2 Gy może być przyczyną zniszczenia 50% oocytów [33]. Pierwotna niewydolność jajników, kiedy pozostaje w nich mniej niż 1000 pęcherzyków, występuje po dawce 6 Gy u osób dorosłych i 10 Gy u dziewcząt leczonych w okresie przedpokwitaniowym i w okresie dojrzewania. Zazwyczaj jednak dawki stosowane w trakcie TBI, wynoszące ≥ 12 Gy, indukują zniszczenie oocytów u wszystkich dziewcząt powyżej 10 r.ż. i u połowy dziewczynek poniżej 10 r.ż. [34]. Stosowanie frakcjonowanych dawek radioterapii aż 5-krotnie zwiększa szansę powrotu funkcji jajników w młodych dziewcząt w porównaniu do pojedynczej dawki napromieniania [35]. Kolejnym czynnikiem uszkadzającym gonady jest busulfan stosowany w wielu protokołach jako element wysokodawkowanej chemioterapii kondycjonującej. W badaniu Legault z roku 1999 aż u 70% dziewcząt i 47% chłopców przygotowywanych do PKK busulfanem i cyklofosfamidem stwierdzano hipogonadyzm [36]. W badaniu Somali z roku 2005, dotyczącym pacjentów po PKK przygotowanych tylko wysokodawkowaną chemioterapią bez radioterapii, hipogonadyzm hipergonadotropowy stwierdzany był u 97% kobiet i 19% mężczyzn [37]. Należy wspomnieć, iż uszkodzenie jajników oraz przewlekła GvHD leczona sterydami wiąże się ściśle z subnormalnym poziomem androgenów u młodych kobiet po PKK [38].
Wszystkie dziewczęta z hipogonadyzmem hipergonadotropowym, u których stwierdza się opóźnienie dojrzewania płciowego (brak rozwoju drugorzędowych cech płciowych przed 13 r.ż.), bądź u których stwierdza się po PKK zatrzymanie rozwoju płciowego (przebieg pokwitania dłuższy niż 5 lat), powinny być leczone od 13 r.ż. stopniowo zwiększanymi dawkami estrogenów celem uzyskania pełnego rozwoju drugorzędowych cech płciowych oraz umożliwienia wystąpienia skoku wzrostowego [5]. Następnie należycie prowadzona zastępcza terapia hormonalna estrogenowo-progesteronowa zapobiega rozwojowi osteoporozy, powikłań sercowo-naczyniowych i demencji [39]. Jest ona również prewencją zaburzeń nastroju, związanych z zaburzeniem libido i poczucia atrakcyjności seksualnej. Ponieważ opisywano spontaniczny powrót prawidłowych cykli menstruacyjnych i prawidłowe ciąże u kobiet z pierwotnie stwierdzonym hipogonadyzmem hipergonadotropowym nawet kilka lat po PKK przeprowadzanym przed 25 r.ż., zaleca się przerwy w stosowaniu estrogenowo-progesteronowej terapii zastępczej na okres około 3–6 miesięcy co 3–4 lata [3]. Możliwość zajścia w ciążę jest bowiem z drugiej strony ograniczona wystąpieniem opisywanej w tej grupie pacjentów przedwczesnej menopauzy. Stwierdzenie wysokich, menopauzalnych poziomów FSH w odstępie przynajmniej jednego miesiąca upoważnia do rozpoznania pierwotnej niewydolności jajników. Poziomy estradiolu bywają bowiem zmienne. Zazwyczaj dla osiągnięcia prawidłowego poziomu estradiolu należy stosować dawkę 100 µg/dobę drogą przezskórną, u niektórych pacjentek dla uzyskania prawidłowych cykli miesięcznych wystarczają dawki mniejsze. Pacjentki z funkcjonalną macicą powinny ponadto otrzymywać preparat progesteronu w drugiej fazie każdego cyklu miesięcznego, np. medroksyprogesteron w dobowej dawce 10 mg przez 12 dni, aby zapobiec hiperplazji endometrium [40]. Można również stosować doustne dwufazowe tabletki koncepcyjne. Droga przezskórna jest jednak preferowana ze względu na mniejsze ryzyko zakrzepicy żylnej. Hormonalna estrogenowo-progesteronowa terapia zastępcza powinna być zakończona, gdy pacjentka osiąga wiek menopauzalny, głównie ze względu na zwiększone ryzyko raka piersi, które jest większe w grupie pacjentek po PKK w porównaniu do ryzyka populacyjnego. Substytucja estrogenowo-progesteronowa jest przeciwwskazana u pacjentek z wywiadem obciążonym udarem mózgu, zakrzepicą żylną, ciężką hipertriglicerydemią, aktywną chorobą wątroby, niezdiagnozowanymi krwawieniami z dróg rodnych oraz występowaniem nowotworów estrogenozależnych, jak np. rak piersi [1].
Zaburzenia metabolizmu kości
Rok do 1,5 roku po leczeniu PKK około 50% pacjentów ma obniżoną gęstość mineralną kości, u co trzeciego pacjenta Z-score wynosi poniżej -1, u 10% Z-score jest < -2,5 i wymagają oni intensywnego leczenia. W związku z obserwowanymi zaburzeniami mineralizacji kośćca u 10% pacjentów po PKK obserwuje się złamania niskoenergetyczne [5, 41]. Dane dotyczące zaburzeń kostnych po przeszczepieniu komórek krwiotwórczych u pacjentów pediatrycznych są stosunkowo skąpe i różnią się nieco od doniesień prezentujących pacjentów dorosłych po PKK. Nysom i wsp. oceniali masę kostną u 25 osób w 8 lat po zakończeniu leczenia onkologicznego obejmującego napromienianie całego ciała oraz allogeniczne przeszczepienie komórek krwiotwórczych. Stwierdzili oni, iż całkowita gęstość mineralna kości oraz gęstość mineralna kości w porównywalnych obszarach u wyleczonych osób jest niższa niż w grupie kontrolnej osób zdrowych [42]. W przeciwieństwie do przytoczonych danych w badaniu Petryka i wsp. u 49 dzieci po PKK prezentowano obniżenie BMD w lędźwiowym odcinku kręgosłupa z -0,56 do -1,1 w 6 miesięcy po transplantacji i do -0,94 w 12 miesięcy po PKK. Jednakże obniżenie całkowitej gęstości mineralnej korelowało tylko w 5,3% z lokalną redukcją masy kostnej w odcinku lędźwiowym kręgosłupa. Liczba pacjentów z osteopenią definiowaną zgodnie w istniejącymi wówczas zaleceniami WHO wzrosła z 18% przed transplantacją do 33% w rok po PKK, w przypadku osteoporozy wartości te zmieniły się z 16% na 19% [43]. W przekrojowych badaniach Bhatia i wsp. również przedstawili redukcję BDM u osób, które przebyły PKK przed 18 r.ż. [44]. Podobnie Taskinen i wsp. prezentowali statystycznie znamienne obniżenie BMD jako późny efekt PKK przeprowadzonego w dzieciństwie aż u 30% badanych pacjentów. Co ciekawe, ich wzrost korelował negatywnie z masą mineralną odcinka lędźwiowego kręgosłupa, wskazując iż BMD nie ma wpływu na wielkość kręgów [45].
Niezależnymi czynnikami ryzyka wystąpienia zaburzeń kostnych po PKK są działania uboczne stosowanych w leczeniu kondycjonującym wysokodawkowanej radioterapii i chemioterapii, a wśród nich uszkodzenie szpiku kostnego biorcy wraz z jego podścieliskiem. Stąd też po PKK dochodzi do względnej przewagi osteoklastów różnicujących się z przeszczepionych komórek dawcy nad osteoblastami pochodzącymi z progenitorów biorcy, uszkodzonymi w przebiegu przygotowania do transplantacji [6]. Ponadto w etiopatogenezie zaburzeń mineralizacji kości po PKK niemałe znaczenie mają też indukowany PKK hypogonadyzm, przewlekła sterydoterapia głównie w leczeniu GVHD oraz stosowanie innych leków immunosupresyjnych, głównie inhibitorów kalineuryny, np. cyklosporyny, jak również długotrwałe unieruchomienie części pacjentów i brak ekspozycji na słońce po PKK.
Z tego powodu w profilaktyce/leczeniu zaburzeń mineralizacji kości istotne znaczenie mają odpowiednia suplementacja witaminy D3 i estrogenowo-progesteronowa hormonalna terapia zastępcza. Wszyscy pacjenci po PKK powinni otrzymywać profilaktyczne dawki wapnia podawanego do posiłku oraz suplementację witaminy D3, tak aby poziom mierzonego jej metabolitu 25-hydroksywitaminy D nie był niższy niż 30 ng/ml. Równie ważne jest wyrównanie zaburzeń funkcji tarczycy i leczenie rhGH osób, u których klinicznie i biochemicznie potwierdzony został jego niedobór. Jednoznacznie udowodnione działanie lecznicze zaburzeń kostnych po PKK mają bisfosfoniany [1, 46], które jednakże nie są zarejestrowane do leczenia dzieci poniżej 18 r.ż.
Podsumowanie
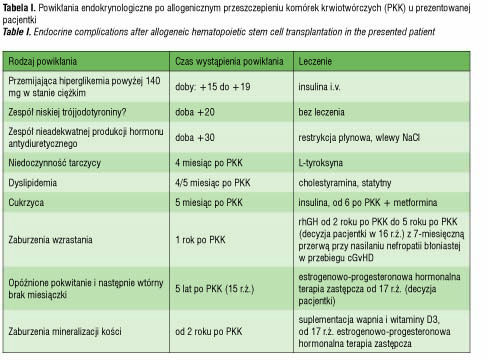
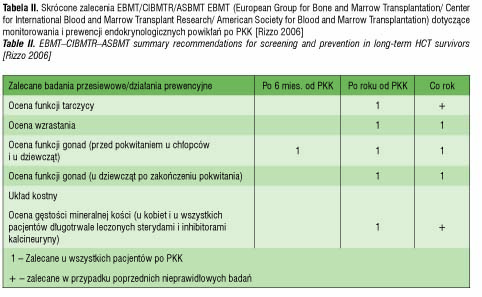
Powikłania endokrynologiczne po PKK (tab. I), które wystąpiły u pacjentki, najpewniej były ubocznym skutkiem radioterapii kondycjonującej oraz działaniami ubocznymi stosowanej farmakoterapii, ze szczególnym uwzględnieniem przewlekłej sterydoterapii w leczeniu choroby przeszczep przeciwko biorcy. Z uwagi na relatywnie duże ryzyko pojawiania się powikłań endokrynologicznych nawet wiele lat po leczeniu PKK konieczny jest jednolity, efektywny program monitoringu tych pacjentów. Zgodnie z propozycją EBMT zaleca się badania obejmujące monitorowanie zaburzeń hormonalnych u pacjentów po PKK (tab. II). Opierając się na naszych doświadczeniach proponowalibyśmy ponadto: badanie hormonów tarczycowych już w trzecim miesiącu po PKK, badanie poziomu HbA1c przynajmniej raz w roku przez kilkanaście lat po PKK.
Piśmiennictwo
1. Savani B.N., Griffith M.L., Jagasia S. et al.; How I treat late effects in adults after allogeneic stem cell complication; Blood 2011:117, 3002-3009
2. Brennan B.M., Shalet S.M.; Endocrine late effects after bone marrow transplant; Br. J. Haematol. 2002:118, 58-66
3. Cohen A., Bekassy A.N., Gaiero A. et al.; Endocrinological late complications after hematopoietic SCT in children; Bone Marrow Transplant. 2008:41, 43-48
4. Shalitin S., Philip M., Stein J. et al.; Endocrine dysfunction and parameters of the metabolic syndrome after bone marrow transplantation during childhood and adolescence; Bone Marrow Transplant. 2006:37, 1109-1117
5. Socie G., Salooja N., Cohen A. et al.; Nonmalignant late effects after allogeneic stem cell transplantation; Blood. 2003:101, 3373-3385
6. Stein E., Ebeling P., Shane E.; Post-transplantation osteoporosis; Endocrinol. Metab. Clin. N. Am. 2007: 36, 937-963
7. Yao S., McCarthy P.L., Dunford L.M. et al.; High prevalence of early-onset osteopenia/osteoporosis after allogeneic stem cell transplantation and improvement after bisphosphonate therapy; Bone Marrow Transplant. 2008:41, 393-398
8. Majhail N.S., Challa T.R., Mulrooney D.A. et al.; Hypertension and diabetes mellitus in adult and pediatric survivors of allogeneic hematopoietic cell transplantation; Biol. Blood Marrow Transplant. 2009:15, 1100-1107
9. Baker K.S., Ness K.K., Weisdorf D. et al.; Late effects in survivors of acute leukemia treated with hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study; Leukemia 2010:24, 2039-2047
10. Sanders J.E., Hoffmeister P.A., Woolfrey A.E. et al.; Thyroid function following hematopoietic cell transplantation in children: 30 years’ experience; Blood 2009:113, 306-308
11. Rizzo J.D., Wingard J.R., Tichelli A. et al.; Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation: join recommendations of the European Group for Blood and Marrow Transplantation, Center for International Blood and Marrow Transplant research, and the American Society for Blood and Marrow Transplantation (EBMT/CIBMTR/ASBMT); Bone Marrow Transplant. 2006:37, 249-261
12. Ranke M.B., Schwarze C.P., Dopfer R. et al.; Late effects after stem cell transplantation (SCT) in children – growth and hormones; Bone Marrow Transplant. 2005:35, 77–81
13. Baker K.S., Ness K.K., Steinberger J. et al.; Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study; Blood 2007:109, 1764-1772
14. Steffens M., Beauloye V., Brichard B. et al.; Endocrine and metabolic disorders in young adult survivors of childhood acute lymphoblastic leukaemia (ALL) or non-Hodgkin lymphoma (NHL); Clin. Endocrinol. 2008: 69, 819-827
15. Hoffmeister P.A., Storer B.E., Sanders J.E.; Diabetes Mellitus in Long-Term Survivors of Pediatric Hematopoietic Cell Transplantation; J. Pediatr. Hematol. Oncol 2004:26, 81-90
16. d’Annunzio G., Bonetti F., Locatelli F. et al.; Insulin resistance in children and adolescents after bone marrow transplantation for malignancies; Haematologica 2006: 10, 1424-1425
17. Bonanomi S., Gaiero A., Masera N. et al.; Distinctive characteristic of diabetes mellitus after hematopoietic cell transplantation during childhood; Pediatr. Transplant. 2006:10, 461-465
18. Tor O., Garg R.K.; Atypical diabetes mellitus associated with bone marrow transplantation; Endocrine Practice 2010: 16, 93-96
19. Traggiai C., Stanhope R., Nussey S. et al.; Diabetes mellitus after bone marrow transplantation during childhood; Med. Pediatr. Oncol. 2002:40, 128-129
20. Tahrani A.A., Cramp C., Moulik P.; The development of non-insulin-dependent diabetes after total body irradiation and bone marrow transplantation in adolescence: a case report and literature review; Pediatric Diabetes 2006:7, 173-175
21. Oh K.W., Lee W.Y., Kang M.I. et al.; Risk Factors of Posttransplant Diabetes Mellitus after Allogeneic Bone Marrow Transplantation; J. Korean Diabetes Assoc. 2000:24, 225-234
22. Taskinen M., Ulla M., Saarinen-Pihkala U.M. et al.; Impaired glucose tolerance and dyslipidaemia as late effects after bone-marrow transplantation in childhood; Lancet 2000: 356, 993-997
23. Lorini R., Cortona L., Scaramuzza A. et al.; Hyperinsulinemia in children and adolescents after bone marrow transplantation; Bone Marrow Transplant. 1995:15, 873-877
24. Hoffmeister P.A., Hingorani S.R., Storer B.E. et al.; Hypertension in long-term survivors of pediatric hematopoietic cell transplantation; Biol. Blood. Marrow Transplant. 2010:16, 515-524
25. Abou-Mourad Y.R., Lau B.C., Barnett M.J. et al.; Long-term outcome after allo-SCT: close follow-up on a large cohort treated with myeloablative regimens; Bone Marrow Transplant. 2010:45, 295-302
26. Gurney J.G., Ness K.K., Sibley S.D. et al.; Metabolic syndrome and growth hormone deficiency in adult survivors of childhood acute lymphoblastic leukemia; Cancer 2006:107, 1303-1312
27. Cohen A., Roveli A., Bakker B. et al.; Final height of patients who underwent bone marrow transplantation for hematological disorders during childhood: a study by the working party for late effects-EBMT; Blood 1999:93, 4109-4115
28. Sanders J.E., Guthrie K.A., Hoffmeister P.A. et al.; Final adult height of patients who received hematopoietic cell transplantation in childhood; Blood 2005:105, 1348-1354
29. Chemaitilly W., Boulad F., Heller G. et al.; Final height in pediatric patients after hyperfractionated total body irradiation and stem cell transplantation; Bone Marrow Transplant. 2007:40, 29–35
30. Holm K., Nysom K., Rasmussen M.H. et al.; Growth, growth hormone and final height after BMT. Possible recovery of irradiation-induced growth hormone insufficiency; Bone Marrow Transplant. 1996:18, 163-170
31. Cohen A., Roveli A., Van-Lint M.T. et al.; Final height of patients who underwent bone marrow transplantation during childhood; Arch. Dis. Child. 1996:74, 437-440
32. Sklar C.A., Mertens A.C., Mitby P. et al.; Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the Childhood Cancer Survivor Study; J. Clin. Endocrinol. Metab. 2002:87, 3136-3141
33. Wallace W.H.B., Thomson A.B., Kelsey T.; The radiosensitivity of the human oocyte; Hum. Reprod. 2003:18, 117-121
34. Sarafoglou K., Boulard F., Gillio A. et al.; Gonadal function after bone marrow transplantation for acute leukemia during childhood; J. Pediatr. 1997:130, 210-216
35. Sanders J.E.; Endocrine problems in children after bone marrow transplant for hematologic malignances; Bone Marrow Transplant. 1991:8, 2-4
36. Legault L., Bonny Y.; Endocrine complications of bone marrow transplantation in children; Pediatr. Transplantation 1999:3, 60-66
37. Somali M., Mpatakoias V., Avramides A. et al.; Function of the hypothalamic-pituitary-gonadal axis in long-term survivors of hematopoietic stem cell transplantation for hematological diseases; Gynecol. Endocrinol. 2005:21, 18-26
38. Hovi L., Saarinen-Pihkala U.M., Taskinen M. et al.; Subnormal androgen levels in young female bone marrow transplant recipients with ovarian dysfunction, chronic GVHD and receiving glucocorticoid therapy; Bone Marrow Transplant. 2004:33, 503-508
39. Piccioni P., Scrirpa P., D’Emilio I. et al.; Hormonal replacement therapy after stem cell transplantation; Maturitas 2004:49, 327-333
40. Nelson L.M.; Clinical practice: primary ovarian insufficiency; N. Engl. J. Med. 2009:360, 606-614
41. Schulte C.M.S., Beelen D.; Bone loss following hematopoietic stem cell transplantation: a long-term follow-up; Blood 2004:103, 3635-3643
42. Nysom K., Holm K., Michaelsen K.F. et al.; Bone mass after allogeneic BMT for childhood leukaemia or lymphoma; Bone Marrow Transplant. 2000:25, 191-196
43. Petryk A., Bergemann T.L., Polga K.M. et al.; Prospective study of changes in bone mineral density and turnover in children after hematopoietic cell transplantation; J. Clin. Endocrinol. Metab. 2006:91, 899-905
44. Bhatia S., Ramsay N.K.C., Weisdorf D. et al.; Bone mineral density in patients undergoing bone marrow transplantation for myeloid malignancies; Bone Marrow Transplant. 1998:22, 87-90
45. Taskinen M., Kananen K., Valimaki M. et al.; Risk factors for reduced areal bone mineral density in young adults with stem cell transplantation in childhood; Pediatr. Transplant. 2006:10, 90-97
46. Tauchmanova L., De Simone G., Musella T. et al.; Effects of various antiresorptive treatments on bone mineral density in hypogonadal young women after allogeneic stem cell transplantation; Bone Marrow Transplant. 2006:37, 81-88








