Endokrynol. Ped. 8/2009;3(28):61-64
DOI: 10.18544/EP-01.08.03.1166
Zespół Kallmanna. Prezentacja przypadku 17-letniej pacjentki
Klinika Endokrynologii i Neurologii Dziecięcej, Uniwersytet Medyczny w Lublinie
Słowa kluczowe: zespół Kallmanna, hipogonadyzm hipogonadotropowy
Streszczenie
Zespół Kallmanna to najczęstsza forma opóźnionego dojrzewania płciowego wtórnego – hipogonadyzmu hipogonadotropowego. Opóźnienie dojrzewania płciowego spowodowane jest zaburzeniami wydzielania gonadotropin, co w efekcie powoduje brak pobudzenia gonad do wydzielania hormonów płciowych. Defekt może mieć różne przyczyny i występować na poziomie przysadki lub podwzgórza. Może być wrodzony, nabyty lub przejściowy
Zespół Kallmana to najczęstsza forma opóźnionego dojrzewania płciowego wtórnego – hipogonadyzmu hipogonadotropowego [1]. Częstość jego występowania wynosi 1:10000 – 1:60000 osób. Opóźnienie dojrzewania płciowego spowodowane jest zaburzeniami wydzielania gonadotropin, co w efekcie powoduje brak pobudzenia gonad do wydzielania hormonów płciowych [2]. Zaburzenia czynności podwzgórza z niedoborem LH-RH i towarzyszącą anosmią są konsekwencją wieloogniskowego uszkodzenia OUN ze współistniejącą aplazją opuszki węchowej.
Zespół Kallmanna stanowi połączenie wrodzonego hipogonadyzmu hipogonadotropowego z brakiem powonienia (anosmia) lub jego upośledzeniem (hiposmia). W rodzinach obciążonych genetycznie występuje zarówno izolowany niedobór gonadotropiny, jak i brak powonienia, podczas gdy u innych izolowany niedobór gonadotropin i normalny zmysł węchu [3]. Po stwierdzeniu hipogonadyzmu hipogonadotropowego dodatni wywiad rodzinny i osłabiony węch są argumentem przemawiającym za diagnozą tego zespołu. Choroba ma charakter rodzinno-dziedziczny i przekazywana jest w sposób autosomalny dominujący lub sprzężony z chromosomem X. Defekt może mieć różne przyczyny, może występować na poziomie przysadki lub podwzgórza, może być wrodzony, nabyty lub przejściowy [4].
Pacjenci z zespołem Kallmanna zgłaszają się z reguły do lekarza z powodu opóźnionego dojrzewania płciowego. U około 80% pacjentów zauważamy brak lub upośledzenie powonienia. U płci męskiej obserwuje się też małe jądra, małe prącie i wnętrostwo, zaś u płci żeńskiej często brak rozwoju gruczołów piersiowych. Do pozostałych objawów należą [5]:
– anomalie w układzie kostnym (zrost palców, skrócony dalszy koniec kości śródręcza, asymetria twarzoczaszki)
– zaburzenia linii środkowej (rozszczep warg lub podniebienia, daltonizm, agenezja nerek, głuchota)
– nieprawidłowości w rotacji trzewi
– wrodzona wada serca
– objawy neurologiczne – ruchy mimowolne, niemożność płynnego śledzenia oczami, dysfunkcja móżdżku).
Opis przypadku
W pracy przedstawiono przypadek 17-letniej pacjentki M.H., urodzonej z C I, P I, prawidł., o czasie. Rozwój w okresie dzieciństwa był prawidłowy. Zgłosiła się do Poradni Endokrynologii z powodu pierwotnego braku miesiączki, opóźnionego dojrzewania płciowego oraz zaburzeń węchu.
Na podstawie badania fizykalnego i wybranych badań diagnostycznych stwierdzono:
– wysoką, szczupłą budowę ciała – 50 kg, 173,5 cm (BMI 17)
– dojrzewanie płciowe M1 P4 A2
– niskie wydzielanie gonadotropin podstawowe i w teście LHRH
– przedpokwitaniowe wartości hormonów płciowych
– niedoczynność tarczycy.
– opóźnienie wieku kostnego o 4 lata.
W badaniu MRI głowy – objawy mikrogruczolaka przysadki.
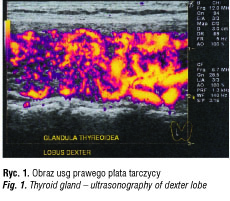
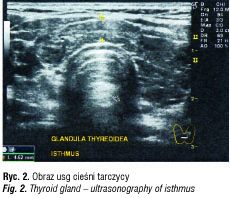
W badaniu usg tarczycy – obraz tarczycy znamienny dla thyreoiditis Hashimoto. Pacjentka zgłaszała również zaburzenia węchu – nie rozpoznawała podstawowych zapachów (ciotka dziewczynki cierpi również na zaburzenia węchu).
Obecnie pacjentka jest pod kontrolą Poradni Endokrynologicznej i Ginekologicznej (przyjmuje Euthyrox i Estrofem).
Wynik badania ultrasonograficznego
Badanie wykonano aparatem LOGIQ 7, linearną sondą matrycową 12 MHz. Gruczoł tarczowy niepowiększony, symetryczny, ale o zdecydowanie nieprawidłowej echostrukturze.
Wymiary prawego płata: a-p 18 mm, med.-lat 24 mm, cr-caud 44 mm (9,5 ml). Wymiary lewego płata: a-p 19 mm, med.-lat 17 mm, cr-caud 46 mm (8 ml). Cieśń około 17,5 ml (norma 13,5 ± 5 ml). Oba płaty i cieśń tarczycy wykazują nieprawidłową, zdecydowanie obniżoną echogeniczność. Echostruktura jest nieregularna, z bardzo licznymi hipoecho/bezechowymi naciekami leukocytarnymi, z pasemkowatymi zwłóknieniami i ewidentnie podwyższoną energią przepływu. Wnioski: obraz tarczycy jest znamienny dla procesu zapalnego (thyreoiditis Hashimoto).
Wynik badania USG narządu rodnego
W badaniu USG przezpochwowo macica w przodozgięciu, w osi długiej 29 mm, w wymiarze strzałkowym w obrębie trzonu 12 mm. Endometrium szczelinowate, o wysokości do 2 mm. Jajnik prawy o wymiarach 30 x 12 mm, z obecnością pojedynczych pęcherzyków o średnicach do 4 mm. Jajnik lewy o wymiarach 34 x 16 mm, największy z kilku pęcherzyków o średnicy 4 mm.
Wynik badania MR
Badanie wykonano aparatem Picker Eclipse 1,5 T, w sekwencjach FAST, FSE, FLAIR, uzyskując obrazy T1- i T2-zależne w projekcjach strzałkowej, czołowej oraz poprzecznej, przed i po dożylnej iniekcji paramagnetycznego środka kontrastowego. W obrazie MR przysadka mózgowa prawidłowej wielkości (12 x 6 mm), symetryczna, z symetryczną szypułą. W obrębie przysadki po stronie prawej widoczne niejednorodne ognisko, odpowiadające mikrogruczolakowi o średnicy 3,5 mm. W jego obrębie widoczne niejednorodne, opóźnione wzmocnienie kontrastowe.
Test LH - RH
LH: 0’ – 0,272; 30‘ – 0,215; 60’ – 6,1; 90’ – 4,44; 120’ – 4,33 U/l
FSH: 0’ – 0,816; 30’ – 1,16; 60’ – 3,39; 90’ – 3,69; 120’ – 3,93 U/l.
Należy zwrócić uwagę na fakt, iż mimo występowania u pacjentki wielu charakterystycznych dla zespołu Kallmanna objawów, rozpoznanie zostało ustalone dopiero po 17 roku życia i w związku z tym późno rozpoczęto terapię hormonalną.
Wydaje się, że w wielu przypadkach opóźnionego dojrzewania płciowego z towarzyszącymi zaburzeniami węchu i występowaniem innych typowych objawów, rozpoznanie tego zespołu nie jest łatwe, a wręcz dość często nie jest on prawidłowo diagnozowany.
Piśmiennictwo
1. Busiah K., Belieu V., Dallot N. et al; Diagnosis of delayed puberty; Arch. Pediatr. 2007, Sep.: 14(9): 1101-1110
2. Antoniazzi F., Zambani G., Tato L.; Delayed puberty; Pediatr. Med. Chir. 1996 Jan.-Feb.: 18(1), 27-31
3. Bhagath B., Layman L.C.; The genetics of hypogonadotropic hypogonadism; Semin. Reprod. Med. 2007 Jul.: 25(4), 276-286
4. Levy C.M., Knudtzon J.; Kallmann syndrome in two sisters with other developmental Anomaliesalso affecting their father; Clin. Genet. 2003 Jan., 43(1), 51-53
5. Hoffman B., Bradshaw K.D.; Delayed puberty and amenorrhea; Semin. Reprod, Med. 2003 Nov., 21(4), 353-362








