Endokrynologia Pediatryczna, Tom 2 | Rok 2003 | Nr 2(3)
DOI: 10.18544/EP-01.02.02.0199
Zaburzenia różnicowania płci o typie odwrócenia płci jako przyczyna opóźnionego pokwitania u młodzieży
1Klinika Endokrynologii Dzieci i Młodzieży Katedry Pediatrii
2Klinika Urologii
3Samodzielna Pracownia Radiologii Collegium Medicum Uniwersytetu Jagiellońskiego w Krakowie
Słowa kluczowe: opóźnione pokwitanie, odwrócenie płci, młodzież
Streszczenie
Retrospektywna analiza przyczyn opóźnionego i/lub nieprawidłowego (heteroseksualnego) pokwitania u 264 pacjentów, spowodowanego pierwotnym uszkodzeniem gonad (hipogonadyzm hipergonadotropowy), wykazała, że jedną z rzadszych przyczyn tych zaburzeń było nieprawidłowe różnicowanie płci o typie odwrócenia płci (sex reversal). Rozpoznanie to ustalono u 7 pacjentów: 3 dziewcząt z czystą dysgenezją gonad 46, XY, 2 pacjentek z częściową dysgenezją gonad 46, XY, 1 pacjentki z zespołem całkowitej niewrażliwości na androgeny oraz u 1 pacjenta, mężczyzny 46, XX. W pracy zwrócono uwagę na możliwość ustalenia rozpoznania u większości chorych dopiero w okresie pokwitania oraz konieczność zwracania uwagi, już od okresu noworodkowego, na cechy zaburzeń rozwoju zewnętrznych narządów płciowych i dysmorfię bodowy ciała, towarzyszące częściowej dysgenezji gonad. Wskazano na podstawowe znaczenie w diagnostyce różnicowej badania fizykalnego, analizy auksologicznej z oceną rozwoju płciowego, badania kariotypu oraz prawidłowej interpretacji uzyskanych wyników badań, opartej na rzetelnej znajomości fizjologii różnicowania płci męskiej. Podkreślono, że w każdym przypadku opóźnionego pokwitania, zarówno u dziewcząt, jak i u chłopców, należy brać pod uwagę zaburzenia różnicowania płci o typie sex reversal
Wstęp
Zaburzenie różnicowania płci polega na niezgodności płci genetycznej i gonadalnej z fenotypem zewnętrznych narządów płciowych. W większości przypadków różnego stopnia obojnacze zewnętrzne narządy płciowe widoczne są u noworodka. Umożliwia to wczesne rozpoznanie zaburzenia różnicowania płci o typie obojnactwa rzekomego (męskiego lub żeńskiego), a także obojnactwa prawdziwego oraz wdrożenie prawidłowego leczenia. W przypadkach ciężkich zaburzeń całkowitego odwrócenia płci (sex reversal) prawidłowy wygląd zewnętrznych narządów płciowych jest przyczyną opóźnienia rozpoznania, zwykle do wieku młodzieńczego, w którym to okresie pokwitanie nie występuje lub pojawiają się nieliczne jego cechy. W niepełnych zespołach odwrócenia płci dyskretne zaburzenia rozwoju zewnętrznych narządów płciowych mogą zostać niezauważone, a rozpoznanie jest stawiane zwykle, gdy wystąpi niezgodne z płcią fenotypową dziecka heteroseksualne pokwitanie. Różnorodność etiologii schorzenia oraz manifestacji klinicznej w okresie pokwitania, a także niedostateczna wiedza lekarzy są częstą przyczyną jeszcze znaczniejszego opóźnienia rozpoznania, nawet do wieku dorosłego. Tak istotne zaburzenie rozwoju płciowego jest przyczyną poważnych urazów psychicznych chorych oraz ich rodzin, a opóźnienie rozpoznania dodatkowo ten uraz pogłębia.
Zaburzenia o typie sex reversal, tak zwana kobieta 46, XY oraz mężczyzna 46, XX, polegają na występowaniu fenotypu przeciwnego do płci genetycznej, określonej wynikiem badania kariotypu. Istnieją różnorodne przyczyny rozwoju fenotypu żeńskiego u osób z kariotypem męskim 46, XY. Należą do nich: czysta dysgenezja gonad 46, XY (complete gonadal dysgenesis 46, XY) [1], zespół całkowitej niewrażliwości na androgeny (complete androgen insensitivity syndrome) [2], a także w niektórych przypadkach częściowa dysgenezja gonad 46, XY, zwana też dysgenetycznym obojnactwem rzekomym męskim (partial gonadal dysgenesis) [3]. U płci genetycznej żeńskiej przyczyną powstawania fenotypu męskiego [4] jest zwykle obecność genu SRY (ang. sex-determining region of theY chromosome, gen zlokalizowany na chromosomie Y determinujący rozwój płci męskiej) na jednym z chromosomów X, spowodowana nieprawidłowym rozdziałem mejotycznym homologicznych chromosomów płciowych (crossing over) – 80% przypadków, a w pozostałych przypadkach – mutacja nieznanych jeszcze genów autosomalnych lub genów chromosomu X [5]. Przyczyną występowania dysgenezji gonad 46, XY jest nieprawidłowa czynność genów tak zwanej kaskady różnicowania płci męskiej: gen SRY, SOX9 (ang. SRY homeobox-like gene 9), WT-1 (ang. Wilms` tumor suppressor gene, gen supresorowy rozwoju guza Wilmsa), SF-1 (ang. steroidogenic factor, czynnik sterydogenezy pierwszy), DAX-1 (ang. dosage-sensitive sex reversal region-adrenal hypoplasia congenita locus on the X chromosome, gene 1) i inne [5–7]. Powodem występowania oporności na androgeny są różnorodne mutacje w obrębie genu dla receptora androgenowego [8].
Materiał i metody
Retrospektywnej ocenie poddano grupę 264 pacjentów (220 dziewcząt oraz 44 chłopców), którzy zgłosili się do Poradni Endokrynologicznej Kliniki Endokrynologii Dzieci i Młodzieży CMUJ w Krakowie w latach 1995-2002 w wieku od 12 do 21 lat z powodu opóźnionego pokwitania, którego przyczyną było pierwotne uszkodzenie gonad (hipogonadyzm hipergonadotropowy). Najczęstszą przyczyną hipogonadyzmu hipergonadotropowego był u tych pacjentów: zespół Turnera, zespół Klinefeltera, zespół dodatkowego chromosomu X lub Y, hipoplazja lub agenezja gonad, stan po obustronnej gonadektomii, stan po radio- i/lub chemioterapii, a także oporność receptorowa na gonadotropiny u chorych z rzekomą niedoczynnością przytarczyc, typ 1a, oraz autoimmunologiczne uszkodzenia gonad u chorych z poliendokrynopatią, typ 1 oraz typ 2. Jedną z rzadszych przyczyn zarówno opóźnienia pokwitania, jak i hipogonadyzmu hipergonadotropowego była ciężka postać zaburzeń różnicowania płci o typie sex reversal, którą stwierdzono u 7 pacjentów (6 dziewcząt, 1 chłopca). Chorych tych poddano szczegółowej analizie, która jest przedmiotem niniejszej pracy.
U każdego pacjenta z opóźnionym pokwitaniem oceniono: stopień rozwoju płciowego według skali Tannera [9], wzrost (3-krotny pomiar wzrostu przy zastosowaniu stadiometrum Harpender), budowę i proporcje ciała, wiek kostny metodą Greulich-Pyle`a [10] oraz podstawowe stężenia hormonu folikulotropowego (FSH), hormonu luteotropowego (LH), estradiolu E2 oraz testosteronu (T) w surowicy krwi. Przy podejrzeniu zaburzenia różnicowania płci wykonano ponadto: oznaczenie kariotypu (preparaty barwione metodą GTG), ocenę ginekologiczną zewnętrznych i wewnętrznych narządów płciowych (u fenotypowych dziewcząt), badanie ultrasonograficzne (USG) miednicy małej z oceną położenia, wielkości i struktury macicy oraz gonad. W surowicy krwi oznaczono stężenia markerów nowotworowych: podjednostki beta choriogonadotropiny kosmówkowej (β-hCG) i alfafetoproteiny (AFP). Badanie histopatologiczne przydatków wykonano u 5 pacjentek, które poddane były gonadektomii. U dwóch pacjentów uzyskano wynik genetycznego badania molekularnego. Wszystkich pacjentów z zaburzeniami różnicowania płci poddano badaniu psychologicznemu.
Cel pracy
1. Analiza etiologii oraz obrazu klinicznego zaburzeń odwrócenia płci, będących przyczyną opóźnionego lub nieprawidłowego pokwitania.
2. Przedstawienie prostych metod diagnostycznych dla ich rozpoznawania w okresie pokwitania.
3. Zwrócenie uwagi na wczesne objawy kliniczne u niektórych chorych z odwróceniem płci, umożliwiające rozpoznanie oraz leczenie przed okresem pokwitania.
Wyniki i dyskusja
Ocena auksologiczna
Wszyscy analizowani pacjenci zgłosili się do Poradni Endokrynologicznej z powodu opóźnionego pokwitania w wieku odpowiednio: 16 4/12 (pacjent 1), 21 1/12 (pacjent 2), 17 2/12 (pacjent 3), 12 3/12 (pacjent 4), 12 1/12 (pacjent 5), 16 8/12 (pacjent 6) oraz 16 4/12 lat (pacjent 7) – tab. I. Pacjentki 4 i 5 zostały skierowane do Poradni Endokrynologicznej z innego ośrodka ze wstępnym rozpoznaniem zespołu niewrażliwości na androgeny. Wszystkie dziewczęta zgłaszały pierwotny brak miesiączki, przy różnym zaawansowaniu innych cech pokwitania, takich jak rozwój piersi i obecność owłosienia łonowego. U pacjentek 1-5 nie obserwowano rozwoju piersi (T I°), obecny był natomiast rozwój owłosienia łonowego (P IV° u pacjentek 1, 2 i 4 oraz P II° u pacjentek 3 i 5). U pacjentki 6 gruczoły piersiowe były rozwinięte w stopniu V wg skali Tannera, ale brak było rozwoju owłosienia łonowego (P I°) oraz w dołach pachowych. Przyczyną zgłoszenia się chłopca była ginekomastia oraz zbyt małe w stosunku do wieku rozmiary jąder (obustronnie 5 ml objętości) oraz prącia (12 cm długości). U pacjentek 1, 2, 5 i 6 stwierdzono prawidłową, kobiecą budowę ciała z prawidłowym lub nadmiernym wzrostem, zwłaszcza u pacjentki 2 (+3,45 SD). U pacjentki 3 widoczna była hiposteniczna budowa ciała oraz liczne cechy dysmorfii twarzy. U pacjentki 4 obecne były cechy budowy ciała charakterystyczne dla zespołu Turnera. U obu pacjentek stwierdzono niedobór wzrostu (odpowiednio: 1,27 SD u pacjentki 3 oraz 1,11 SD u pacjentki 4), a także umiarkowany niedorozwój umysłowy z zaburzeniem sfery emocjonalnej, który stwierdzono w badaniu psychologicznym. U pacjenta 7 ginekomastia współistniała z ginekoidalną budową ciała. Wiek kostny u pacjentek 1, 2 i 3 był znacznie opóźniony, a u pozostałych badanych odpowiadał wiekowi chronologicznemu.
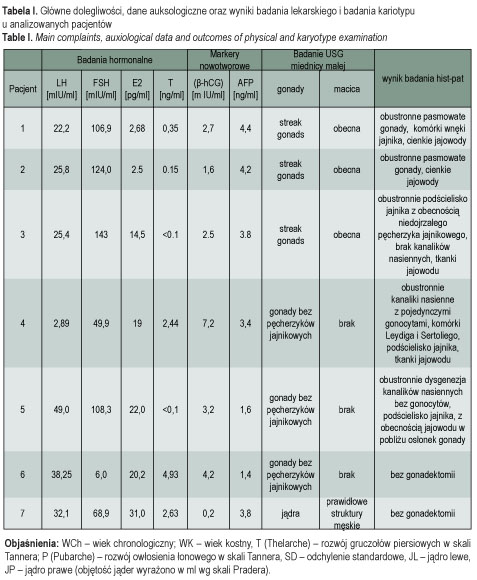
Badania ginekologiczne oraz ultrasonograficzne
W badaniu ginekologicznym u pacjentek 1-3 oraz 5 stwierdzono prawidłową budowę żeńskich zewnętrznych narządów płciowych. U pacjentki 4 występowały cechy wirylizacji zewnętrznych narządów płciowych (przerośnięta łechtaczka) oraz hirsutyzm, a także drożne, ale zwężone ujście pochwy. Badanie ginekologiczne pacjentki 6 wykazało żeńskie zewnętrzne narządy płciowe, brak błony dziewiczej, a w miejscu pochwy szeroki, ślepo zakończony zachyłek długości około 5 cm. Badaniem ginekologicznym oraz USG miednicy małej wykazano obecność macicy u pacjentek 1-3 oraz jej brak u pacjentek 4-6 (tab. II). U wszystkich pacjentek uwidoczniono zlokalizowane śródbrzusznie homogenne gonady bez pęcherzyków jajnikowych. Badaniem USG u pacjenta 7 wykazano obecność obu jąder o prawidłowym echu zlokalizowanych w worku mosznowym.
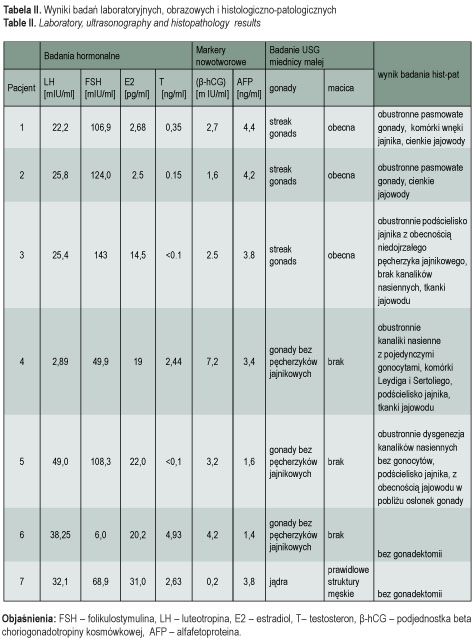
Badania hormonalne
U wszystkich pacjentów stwierdzono nieprawidłowe, podwyższone stężenia obu lub jednej z gonadotropin (FSH i/lub LH) w surowicy krwi, przy obniżonych wartościach estradiolu u pacjentek z fenotypem żeńskim i obniżonym stężeniu testosteronu u pacjenta z fenotypem męskim (2,6 ng/ml) – tab. II. Wysokie stężenia FSH, wskazujące na dysgenezję gonad, stwierdzono u pacjentek 1-5, a także u pacjenta 7. U pacjentki 6 zwracało uwagę podwyższone stężenie LH (38 mIU/ml) oraz wysokie stężenie testosteronu (4,93 ng/ml).
Na podstawie przedstawionego obrazu klinicznego i wyników badań ustalono następujące rozpoznania: czystą dysgenezję gonad 46, XY (pacjentki 1-3), częściową dysgenezję gonad 46, XY (pacjentki 4 i 5), zespół całkowitej niewrażliwości na androgeny (pacjentka 6), mężczyzna 46, XX (pacjent 7).
U wszystkich badanych oznaczono markery nowotworowe: β-hCG oraz AFP (tab. II) z powodu ponad 30% ryzyka procesu nowotworowego o typie gonadoblastoma i/lub germinoma (dysgerminoma/seminoma) w dysgenetycznych gonadach [1] u pacjentek 1-5 oraz podwyższonego ryzyka u pacjentów pozostałych. Tylko u pacjentki 4 stężenie β-hCG było nieznacznie podwyższone i mogło sugerować przemianę nowotworową gonady/gonad. Z podanych powodów zabieg usunięcia gonad (gonadektomia) został wykonany u pacjentek 1-5. Pacjentka 4 wymagała ponadto plastyki przerośniętej łechtaczki, a pacjent 7 (mężczyzna 46, XX) – chirurgicznej korekcji ginekomastii. Rodzice pacjentki 6, z zespołem całkowitej niewrażliwości na androgeny, poinformowani o istocie choroby i konieczności wykonania gonadektomii, nie wyrazili zgody na zabieg.
Badanie histopatologiczne
Badania histologiczno-patologiczne gonad wykazały obustronnie pasmowate gonady (streak gonads) u pacjentek 1 i 2. U pacjentki 3 stwierdzono obecność pojedynczych niedojrzałych pęcherzyków jajnikowych w podścielisku jajnika, bez obecności kanalików nasiennych oraz komórek Leydiga (ryc. 1). U pacjentki 4 uwidoczniono w gonadach podścielisko jajnika z obecnymi kanalikami nasiennymi z komórkami Sertoliego oraz pojedynczymi komórkami plemnikotwórczymi (gonocytami), z zachowanym częściowo systemem komórek Leydiga (ryc. 2). U pacjentki 5 stwierdzono w podścielisku jajnika dysgenetyczne kanaliki nasienne bez gonocytów oraz brak komórek Leydiga. U wszystkich pacjentek obecne były tkanki jajowodów w postaci obustronnie cienkich jajowodów (pacjentki 1, 2 i 5) lub tkanek jajowodów w styczności z gonadą (pacjentki 3 oraz 4).
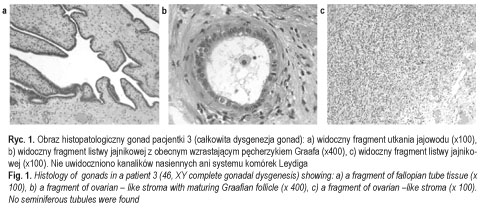
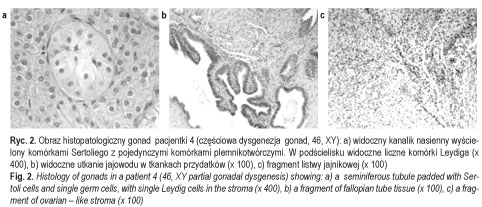
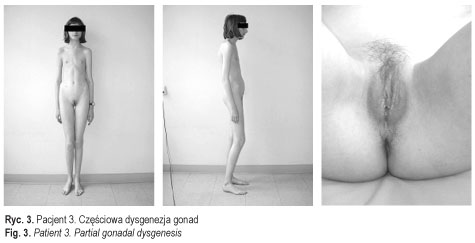
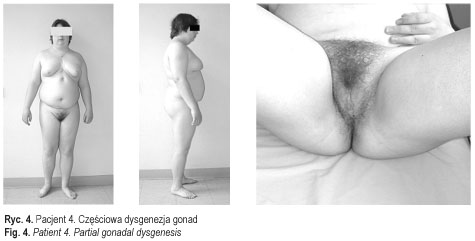
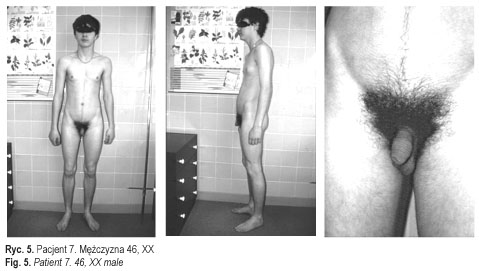
Dane te potwierdziły rozpoznanie całkowitej dysgenezji gonad u pacjentek 1-3 (ryc. 3 przedstawia pacjentkę 3) oraz częściowej dysgenezji gonad u pacjentek 4-5 [11] (ryc. 4). U chorych operowanych wykluczono carcinoma in situ. U wszystkich chorych, oprócz pacjentki 6, włączono leczenie substytucyjne hormonami płciowymi zgodnie z płcią fenotypową. Postępowanie to miało na celu wykształcenie i utrzymanie feminizacji u dziewcząt i maskulinizacji u chłopca, i było jednocześnie profilaktyką osteoporozy. Pacjent 7 (ryc. 5) pozostaje w stałej obserwacji struktury gonad. Rodzice pacjentki 6 zmienili miejsce zamieszkania. Pacjenci i ich rodziny korzystali ze wsparcia psychologicznego, które jest integralną częścią procesu terapeutycznego u chorych z zaburzeniami różnicowania płci. Ustalono zgodną z płcią fenotypową orientację i identyfikację płci u wszystkich pacjentów. Pacjent 7 (mężczyzna 46, XX) został zwolniony ze służby wojskowej.
Ważnym zagadnieniem jest możliwość posiadania przez tych chorych potomstwa. U dziewcząt 1-3, u których stwierdzono prawidłowo zbudowaną macicę, istnieje taka możliwość przez wszczepienie zapłodnionego pozaustrojowo oocyta dawcy. U pozostałych pacjentek oraz u mężczyzny 46, XX posiadanie potomstwa jest możliwe w wyniku adopcji.
Diagnostyka różnicowa
Rozpoznanie czystej dysgenezji gonad 46, XY u pacjentek 1, 2 i 3 potwierdzały następujące dane: brak zależnych od estrogenów cech pokwitania (rozwoju piersi i miesiączki), przy prawidłowym rozwoju zewnętrznych i wewnętrznych narządów płciowych żeńskich oraz obecności rozwoju owłosienia łonowego. Na podstawie wiedzy o etapach różnicowania płci męskiej można wnioskować, że: obecność pochodnych przewodów Müllera (macica, 1/3 górnej części pochwy) przy męskim kariotypie 46, XY wyklucza czynność hormonu antymüllerowskiego (MIH – müllerian ihibiting hormone) wydzielanego przez komórki Sertoliego jąder płodowych. Natomiast brak pochodnych przewodów Wollfa (nasieniowody, najądrza) i prawidłowe żeńskie zewnętrzne narządy płciowe wykluczają zarówno lokalną, jak i systemową aktywność testosteronu, produkowanego przez komórki Leydiga w tych jądrach. Dane te wskazują na głębokie zaburzenie na etapie różnicowania pierwotnej gonady w jądro i brak jego hormonalnej czynności w okresie 8-14 tygodnia ciąży, co było zgodne z wykazaniem pasmowatej tkanki łącznej z lub bez podścieliska jajnika w miejscu typowym dla jajników (streak gonads). Za brakiem czynności hormonalnej gonad przemawiały również kliniczne i hormonalne cechy hipogonadyzmu hipergonadotropowego. Brak determinacji i rozwoju jądra, przy obecności w kariotypie chromosomu Y, wskazują na brak aktywności w życiu płodowym czynnika determinacji rozwoju jąder (TDF – testis determining factor), zależnego od produktu polipeptydowego genu SRY, sugerując mutację tego genu lub innych genów tak zwanej kaskady różnicowania płci męskiej. Mutację genu SRY wykrywa się u około 15% pacjentek z czystą dysgenezją gonad 46, XY [12]. U pacjentki 1 analizą objęto fragment genu SRY (609 pz), który odpowiedzialny jest za różnicowanie się pragonady w jądro [13]. Uzyskany wynik SSCP (single-stranded conformation polymorphysm) nie wskazywał jednak na obecność mutacji w badanym fragmencie genomu, kodującym konserwatywny region odpowiedzialny za łączenie się z DNA. Odczytane sekwencje były zgodne z sekwencją podawaną za prawidłową w Gene Bank zarówno u chorej, jak i u członków jej rodziny. Prawdopodobną przyczyną zaburzeń u tej pacjentki oraz pacjentek 2 i 3 mogła być nieprawidłowa czynność innych genów, takich jak: WT-1, SOX-9, DAX-1, SF-1, lub genów zlokalizowanych w rejonach 9p, 2q, 10q [5,14]. Za powstanie czystej dysgenezji gonad w 30% przypadków odpowiedzialna jest nieprawidłowa wymiana pomiędzy homologicznymi genami PRKX, PRKY (protein kinase gene) regionu pseudoautosomalnego chromosomów X i Y [15]. U pacjentki 3 (czysta dysgenezja gonad 46, XY) nie uzyskano jeszcze wyniku badania genetycznego, jednak stwierdzone u niej cechy dysmorfii twarzy, hiposteniczna budowa ciała, niski wzrost i opóźniony rozwój umysłowy, odpowiadają przypadkom mikroduplikacji fragmentu ramienia krótkiego chromosomu X, opisanym przez Scherera: Xp21.2-22.2 lub 21.2-22.3 [16] oraz Sterna: Xp21-22.3 [17]. Zaburzenia te powinny skłaniać do wykonania u tej pacjentki oznaczenia kariotypu we wcześniejszych okresach życia, jeszcze przed wystąpieniem zaburzeń pokwitania.
U pacjentek 4 i 5 w okresie noworodkowym przyjęto w innym ośrodku błędne rozpoznanie zespołu niewrażliwości na androgeny na podstawie stwierdzenia nieznacznego powiększenia łechtaczki przy prawidłowym rozwoju zewnętrznych narządów płciowych, braku macicy oraz stwierdzenia kariotypu 46, XY. Zarówno cechy obojnactwa (jakkolwiek dyskretnie zaznaczone), ale także obecne u pacjentki 4 fenotyp turnerowski (występujący u około 25% chorych), niedobór wzrostu, nieznaczne opóźnienie rozwoju psychicznego oraz współistniejące wady innych układów (obustronne przepukliny pachwinowe, odpływy pęcherzowo-moczowodowe III stopnia, a także operowane w okresie niemowlęcym: ubytek międzyprzedsionkowy typu drugiego oraz zastawkowe zwężenie aorty) sugerowały raczej rozpoznanie częściowej dysgenezji gonad. Taki objaw, jak stwierdzony u obu pacjentek brak macicy, który jest charakterystyczny dla zespołu niewrażliwości na androgeny i tylko sporadycznie występuje w częściowej dysgenezji gonad [18], nie mógł być kryterium decydującym do rozpoznania. Nie wykonano jednak koniecznej w tych przypadkach diagnostyki różnicowej dla ustalenia przyczyn obojnactwa rzekomego męskiego. Przyjmując rozpoznanie zespołu niewrażliwości na androgeny, nie wykonano również gonadektomii, co miało na celu zapewnienie prawidłowej feminizacji rozwoju żeńskich zewnętrznych narządów płciowych (aromatyzacja „niewykorzystanego” testosteronu do estradiolu). Przyczyną skierowania do Kliniki Endokrynologii był nieoczekiwany u obu pacjentek brak rozwoju gruczołów piersiowych w okresie pokwitania oraz wirylizacja ciała i zewnętrznych narządów płciowych u pacjentki 4 (pokwitanie heteroseksualne). Ustalone w Klinice rozpoznanie częściowej dysgenezji gonad było zgodne z wykazanym, za pomocą oznaczeń gonadotropin, pierwotnym uszkodzeniem gonad (hipogonadyzmem hipergonadotropowym) oraz wynikiem badania histologicznego gonad u obu pacjentek. Stężenie testosteronu, wystarczające do wywołania wirylizacji w okresie pokwitania u pacjentki 4, świadczyło o zachowanej częściowo czynności systemu komórek Leydiga. Wykształcenie płodowego jądra, jakkolwiek dysgenetycznego, przemawiało za innym niż w czystej dyzgenezji gonad 46, XY zaburzeniem determinacji płci męskiej. Zaburzenie to dotyczy prawdopodobnie późniejszych etapów kaskady różnicowania płci męskiej.
U pacjentki 6 rozpoznano zespół całkowitej niewrażliwości na androgeny. Za takim rozpoznaniem u chorych przemawiała prawidłowa, żeńska budowa ciała, żeńskie zewnętrzne narządy płciowe, brak owłosienia łonowego, ślepo zakończony zachyłek pochwy, brak macicy i pierwotny brak miesiączki oraz prawidłowy rozwój piersi w okresie pokwitania (P V°). Podstawą rozpoznania było stwierdzenie u pacjentki w okresie pokwitania wysokiego stężenia testosteronu w surowicy krwi (4,93 ng/ml), co było klinicznym dowodem całkowitego braku jego aktywności. Na podstawie wiedzy o etapach różnicowania płci męskiej można było wnioskować, że: brak pochodnych przewodów Müllera (macica, 1/3 górnej części pochwy), przy męskim kariotypie 46, XY, wskazuje na zachowaną czynność MIH powstającego w jądrach płodowych. Obecność żeńskich zewnętrznych narządów płciowych wskazuje na brak aktywności testosteronu w krążeniu systemowym w okresie płodowym. Brak owłosienia łonowego w wieku 16 lat informował nie tylko o braku aktywności znacznie podwyższonych stężeń testosteronu, ale także braku efektu działania androgenów nadnerczowych. Dane te świadczą o prawidłowym różnicowaniu jąder w życiu płodowym i wskazują na całkowitą nieaktywność receptora androgennego u obu pacjentek. Mechanizm ten pozostaje w zgodności z brakiem cech hipogonadyzmu hipergonadotropowego (prawidłowe stężenia FSH – 6,0 mIU/l), przy charakterystycznym dla tego zespołu podwyższonym stężeniu LH (38,25 mIU/l). Prawidłowy rozwój piersi oraz feminizacja sylwetki pacjentki 6 były wynikiem aromatyzacji testosteronu do estradiolu, w stężeniu charakterystycznym dla płci żeńskiej (20,2 pg/ml), oraz androgenów nadnerczowych do estronu.
U pacjenta 7 rozpoznanie zespołu mężczyzny 46, XX sugerowały: brak postępu pokwitania zgodny z małymi rozmiarami jąder (obustronnie 5 ml) i prącia (długości 12 cm) oraz hormonalne (podwyższone FSH, niskie stężenie testosteronu) i kliniczne cechy hipogonadyzmu (ginekoidalna budowa ciała i ginekomastia). Podstawą rozpoznania było stwierdzenie kariotypu 46, XX, niezgodnego z fenotypową płcią męską. Obecność u chłopca struktur pochodzących z przewodów Wollfa oraz męski fenotyp zewnętrznych narządów płciowych wskazywały na aktywność testosteronu, zarówno produkowanego lokalnie przez komórki Leydiga, jak i działającego systemowo, co razem z brakiem struktur pochodzenia müllerowskiego (prawidłowa czynność MIH) wskazywało na istnienie zróżnicowanego jądra w życiu płodowym. Było to dowodem na dostatecznie skuteczne działanie TDF na etapie różnicowania pierwotnych gonad, a tym samym na obecność w genotypie genu SRY. Wynik badania metodą PCR (polymerase chain reaction) potwierdził u pacjenta 7 obecność dwóch sekwencji chromosomu Y: PABY i SRY, których prawidłową lokalizacją są odpowiednio Yp11.32 i Yp11.3. Tłumaczy to rozwój jąder i fenotypu męskiego u osoby z kariotypem żeńskim. Jednak stwierdzone u chłopca kliniczne objawy hipogonadyzmu o charakterze hipergonadotropowym (T-2.63 ng/ml, FSH-68,9 mIU/l) wskazywały na niepełny rozwój tych jąder (dysgenezja jąder). Ginekomastia była spowodowana obwodową aromatyzacją testosteronu do estrogenów i nieprawidłową proporcją obu hormonów (T/E2).
Wnioski
1. W każdym przypadku opóźnionego i/lub nieprawidłowego pokwitania, zarówno u dziewcząt, jak i u chłopców, należy brać pod uwagę zaburzenia różnicowania płci o typie odwrócenia płci.
2. Dla rozpoznania w okresie pokwitania wystarczające są proste badania auksologiczne, oznaczenie podstawowych stężeń FSH, LH, T, E2 oraz kariotypu, pod warunkiem znajomości fizjologii różnicowania płci męskiej.
3. W niektórych przypadkach odwrócenia płci (zarówno czystej, jak i mieszanej dysgenezji gonad) istnieją objawy kliniczne, które powinny prowadzić do rozpoznania oraz leczenia (gonadektomii) przed okresem pokwitania.
Piśmiennictwo
1. Berkovitz G.D., Fechner P.Y., Zacur H. W. et al.; Clinical and pathologic spectrum of 46, XY gonadal dysgenesis: Its relevance to the understanding of sex differentiation; Medicine 1991:70, 375
2. Migeon C.J., Brown T.R., Fichman K.R.; Androgen insensitivity syndrome. [w:] Pediatric and Adolescent Endocrinology: The intersex child; Red. Josso N. S. Karger Paris 1981:8, 171
3. Migeon C.J.; Male pseudohermaphroditismus; Annales d’Endocrinologie (Paris) 1980:41, 311
4. De la Chapelle A.; Nature and origin of males with XX sex chromosomes; Am. J. Med. Genet. 1972:24, 71
5. Sarafoglou K., Ostrer H.; Familial Sex Reversal: A Review; J. Clinic. Endocrinol. Metab. 2000:85 (2), 483
6. Bilbao J.R., Loridan L., Castano L.; A novel post zygotic nonsense mutation in SRY in familial XY gonadal dysgenesis; Hum. Genet. 1996:97, 537
7. Veitia R., Ion A., Bardaux S., Jobling M.A. et al.; Mutation and sequence variants in the testis-determining region of the Y chromosome in individuals with a 46, XY female phenotype; Hum. Genet. 1996:99, 648
8. Ahmed S.F., Cheng A., Dovey L. et al.; Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome; J. Clinic. Endocrinol. Metab. 2000:85, 658
9. Tanner J.M., Whitehouse R.H., Takaishi M.; Standards from birth to maturity for height, weight, height velocity, and weight velocity: British children, 1965; Part I. Arch. Dis. Child. 1996:41, 454
10. Greulich W.W., Pyle I.; Radiographic atlas of skeletal development of the hand and wrist; Stanford University Press 1959
11. Radakovic B., Julic S., Bukoviec D. et al.; Morphology of gonads in pure XY gonadal dysgenesis; Coll. Anthropol. 1999:23, 203
12. Hawkins J.R., Taylor A., Goodfellow P.N. et al.; Evidence for increased prevalence of SRY mutation in XY females with complete rather than partial gonadal dysgenesis; Am. J. Hum. Genet. 1992:51, 979
13. Sinclair A.H., Berta P., Palmer M.S. et al.; A gene from the human sex determination region encodes a protein with homology to a conserved DNA-binding motif; Nature 1990:346, 240
14. McDonald M.T., Flejter W., Sheldon S. et al.; XY sex reversal and gonadal dysgenesis due to 9p24 monosomy; Am. J. Med. Genet. 1997:73, 321
15. Schiebel K., Winkelmann M., Mertz A. et al.; Abnormal XY interchange between a novel isolated protein kinase gene. PRKY, and its homologue, PRKX accounts for one third of all (Y+) XX males and (Y-) XY females; Hum. Mol. Genet. 1997:6, 1985
16. Scherer G., Schempp W., Baccichetti C. et al.; Duplication of an Xp segment that includes the ZFX locus causes sex inversion in men; Hum. Genet. 1989:81, 291
17. Stern H.J., Garrity A.M., Saal H.M. et al.; Duplication of Xp21 and sex reversal: Insight into mechanism of sex determination; Am. J. Hum. Genet. 47
18. Fechner P.Y., Marcantonio S.M., Ogata T. et al.; Report of a Kindred with X-Linked (or Autosomal Dominant Sex-Limited) 46, XY Partial Gonadal Dysgenesis; J. Clin. Endocrinol. Metab. 1993:76, 1248








