Endokrynologia Pediatryczna, Tom 2 | Rok 2003 | Nr 1(2)
DOI: 10.18544/EP-01.02.01.1044
Rodzinne występowanie raka tarczycy
Klinika Endokrynologii i Diabetologii Wieku Rozwojowego Instytutu Pediatrii AM w Poznaniu
Słowa kluczowe: rak tarczycy, predyspozycja genetyczna, dzieci
Streszczenie
Zdecydowana większość przypadków raka tarczycy ma charakter sporadyczny. Postać dziedziczna raka występuje najczęściej w grupie raka rdzeniastego (25%). Tylko 5% przypadków raka brodawkowatego ma charakter rodzinny, a inne typy raka tarczycy (pęcherzykowy, wyspowy, anaplastyczny) jeszcze rzadziej występują rodzinnie. W pracy opisano aktualny pogląd na temat rodzinnej predyspozycji do wystąpienia raka tarczycy. Znajomość tego zagadnienia, a szczególnie współwystępowania raka tarczycy w zespołach mnogich nowotworów może pozwolić na szybkie wykrycie poszczególnych nowotworów i określić zagrożenie nowotworem u członków ich rodzin.
Wstęp
Guzki tarczycy, dość powszechnie występując u dorosłych, stanowią trudny problem diagnostyczny, jeśli są wykryte u dzieci. Jednak u dzieci łatwiej wybrać najlepszy sposób postępowania w związku z tym, że guzy te są najczęściej nowotworami, łagodnymi bądź złośliwymi. Guzy nowotworowe wymagają leczenia operacyjnego i następowego leczenia ukierunkowanego w zależności od ostatecznego wyniku badania histopatologicznego. Obciążony wywiad rodzinny w zakresie chorób tarczycy i / lub chorób nowotworowych zajmuje istotne miejsce w algorytmie postępowania diagnostyczno-lecz niczego i wpływa na wybór metody leczenia. Każ dorazowo pojawiają się pytania: czy obecność guzka nowotworowego u dziecka ma charakter sporadyczny, czy jest uwarunkowana rodzinnie oraz czy rak tarczycy jest jedynym nowotworem, czy też stanowi element zespołu chorobowego np. zespołu mnogiej gruczolakowatości wewnątrzwydzielniczej MEN 2A lub MEN 2B. O ile w grupie wszystkich raków tarczycy około 5-10% uważa się za postaci dziedziczne, to w grupie raka rdzeniastego tarczycy ten odsetek sięga nawet 25% [1–2].
Rak rdzeniasty tarczycy
Rak rdzeniasty tarczycy (MTC) wywodzi się z komórek okołopęcherzykowych C tarczycy i jest najczęstszym dziedzicznym nowotworem tarczycy. Komórki C tarczycy produkują i wydzielają w nadmiarze kalcytoniny. Kalcytonina była przez lata uznawana za istotnego antagonistę działania parathormonu, hormonu wydzielanego przez przytarczyce, jednak obecnie uważa się, że jej fizjologiczne znaczenie u dorosłych pozostaje niewielkie. Podwyższone stężenie kalcytoniny jest bardzo czułym i specyficznym markerem MTC. W diagnostyce różnicowej podwyższonego stężenia kalcytoniny (norma < 10 pg/ml) uwzględnić należy stany następujące: (1) ciążę, (2) stosowanie leków antykoncepcyjnych, (3) niewydolność nerek, (4) choroby wątroby i (5) różne guzy nowotworowe. 3-krotny wzrost stężenia kalcytoniny (w stosunku do stężenia podstawowego) w teście z pentagastryną jest uznany za wystarczający do podejrzewania MTC. W przypadku guzów zaawansowanych może występować odróżnicowanie się guza z następowym obniżeniem produkcji i wydzielania kalcytoniny. Wówczas markerem tak zaawansowanej postaci MTC jest antygen karcinoembrionalny (CEA) – norma do 10 ng/ml [3].
Częstość występowania MTC waha się w granicach 5-10% ogółu przypadków raka, w analizie zaś obejmującej dzieci i młodzież z chorobą guzkową tarczycy poniżej 19 roku życia w regionie wielkopolskim w latach 1996-2000 zanotowano 1 przypadek, co stanowiło 2,7% wszystkich przypadków raka [4].
Uważa się, że tylko 25% wszystkich przypadków MTC ma podłoże dziedziczne (hMTC), a pozostałe 75% to przypadki sporadyczne [1–3]. Postaci rodzinne MTC występują w następujących formach:
1. Izolowana rodzinna postać MTC (FMTC),
2. Zespół mnogiej gruczolakowatości wewnątrzwydzielniczej (Multiple Endocrine Neoplasia):
a) MEN 2A (zespół Sipple’a), b) MEN 2B.
Istnieją warianty zespołu MEN2A, w których dodatkowo występuje liszaj amyloidowy skóry (MEN2A/CLA) oraz choroba Hirschprunga (MEN2A/HD). Według niektórych autorów wariantami zespołu MEN2A są FMTC, a także rodzinnie występujący guz chromochłonny [5].
W przypadkach sporadycznych FMTC i MEN 2A mogą nie być rozpoznawane w okresie dziecięco-młodzieżowym. Z kolei, MEN 2B jest możliwy do wykrycia w każdym wieku, nawet bezpośrednio po urodzeniu, gdyż zmiany o charakterze zwojakonerwiaków języka i warg mogą być już wówczas stwierdzone (obserwacje własne). Również bardzo wcześnie, tj. ok. 2 roku życia, można stwierdzić zmiany okulistyczne, jak: pogrubienie nerwów rogówki, nerwiaki spojówki czy poszerzenie naczyń spojówkowych przy rąbku rogówki [6]. Nerwiaki w śluzówce przewodu pokarmowego mogą również wyprzedzać o wiele lat pojawienie się ogniska MTC. Objawy związane z ich występowaniem mogą być niecharakterystyczne i obejmować: zaparcia, biegunki, niedobór masy ciała, ból brzucha, trudności w połykaniu. Część dzieci mogła być nawet operowana z powodu ww. objawów. Znajomość fuch faktów pozwala na szybkie wykrycie zagrożenia rozwoju MTC i podjęcie właściwego postępowania (profilaktyczna tyreoidektomia) [5]. Charakterystyka poszczególnych postaci dziedzicznego raka rdzeniastego tarczycy została przedstawiona w tabeli I.
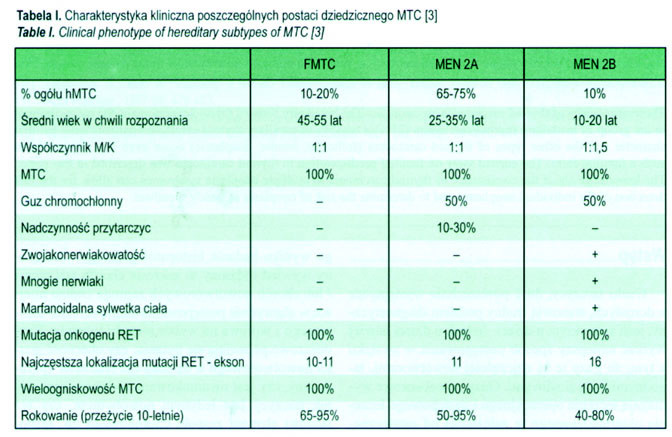
Dzięki zastosowaniu metod biologii molekularnej obecnie w więcej niż 95% przypadków dziedzicznego MTC stwierdza się mutacje onkogenu RET w linii germinatywnej [7–8]. Gen zlokalizowany jest na chromosomie 10, a jego mutacje mają charakter mutacji aktywnych (garo of function) i dziedziczone są w sposób autosomalny dominujący [9–10]. Onkogen RET koduje receptor kinazy tyrozynowej i ulega ekspresji w tkankach pochodzących z grzebieni nerwowych, włączając komórki układu nerwowego ośrodkowego i obwodowego oraz tkanki neuroendokrynowe. Prawie wszyscy pacjenci omawiani w literaturze, u których wykonano zabieg profilaktycznej tyreoidektomii, prezentowali morfologiczne zmiany w obrębie komórek C tarczycy. Zmiany te określono jako hiperplazję komórek C, uważaną za stan pre-MTC; występowały też ogniska MTC, którym mogły towarzyszyć dodatkowo zmiany przerzutowe w węzłach. Ryzyko wystąpienia przerzutów do węzłów chłonnych szyjnych bocznych wzrasta znacząco powyżej 15 roku życia i dlatego poza tym wiekiem limfadenektomia jest wykonywana rutynowo u każdego pacjenta z MTC. MTC towarzyszący zespołowi MEN 2B cechuje się największą agresywnością. stąd tak ważna jest u tych pacjentów wczesna diagnoza i leczenie już w pierwszym roku życia (całkowita tyreoidektomia włącznie z obustronną limfadenektomią), co istotnie poprawia rokowanie u tych pacjentów. Zdecydowana większość (95%) pacjentów z zespołem MEN 2B jest nosicielem mutacji w eksonie 16 (kodon 918), podobnie jak w przypadku sporadycznego MTC [5]. Izolowany zespół nerwiaków śluzówki nie ujawnił ww. mutacji i stąd taką izolowaną postać zaburzeń trudno zaliczyć do nietypowej postaci zespołu MEN 2B [11]. Za jego wystąpienie są odpowiedzialne prawdopodobnie inne geny aniżeli onkogen RET. Z kolei w zespole MEN 2A najczęściej występuje mutacja w eksonie 11 (kodon 634) – 85% [5]. Lokalizację nieprawidłowości genetycznych onkogenu RET w poszczególnych postaciach klinicznych zespołu mnogiej gruczolakowatości przedstawiono na rycinie 1.
Badania diagnostyczne:
• Badania hormonalne: kalcytonina (ew. test stymulacyjny z pentagastryną u nosicieli mutacji onkogenu RET), PTH, ACTH, FT3, FT4, TSH.
• Wykładniki gospodarki Ca-P: wapń, fosfor, magnez, fosfataza zasadowa.
• Markery nowotworowe: CEA.
• Dobowa zbiórka moczu na zawartość VMA i dopaminy.
• Wielokrotne pomiary ciśnienia tętniczego.
• Badania obrazowe: USG szyi i jamy brzusznej, KT lub MR szyi, klatki piersiowej – dla miejscowej i odległej oceny stopnia inwazji raka tarczycy (przerzuty); rozważyć badanie scyntygraficzne tarczycy.
• MR klatki piersiowej i nadnerczy – dla poszukiwania ew. rozrostu tkanki chromochłonnej (85-90% lokalizacja nadnerczowa, 10-15% lokalizacja poznadnerczowa).
• Biopsja aspiracyjna cienkoigłowa tarczycy (w przypadku obecności guzka tarczycy).
• Analiza genu RET [12–14].
• Badanie okulistyczne.
Postępowanie lecznicze:
1. Rodzaj zabiegu – całkowita tyreoidektomia z wycięciem węzłów chłonnych szyjnych środkowych (w każdym przypadku MTC o lokalizacji tarczycowej także bez obecności miejscowych przerzutów węzłowych).
2. Czas wykonania profilaktycznej tyreoidektomia: 1 rok życia – MEN 2B,
5-6 rok życia – MEN 2A i FMTC.
3. Konieczne jest potwierdzenie lub wykluczenie współistnienia guza chromochłonnego w nadnerczu(ach) lub o innej lokalizacji. Guz chromochłonny należy usunąć wcześniej aniżeli guz tarczycy, gdyż nieoczekiwany nadmiar uwalnianych katecholamin może spowodować stan zagrożenia życia w trakcie zabiegu operacyjnego guza tarczycy. Może być to pierwsza manifestacja kliniczna obecności guza chromochłonnego. Guz chromochłonny występuje w obu nadnerczach w 50-80%.
4. Pooperacyjne leczenie zastępcze L-tyroksyną – TSH w przedziale 1,0-2,0 µIU/mL (nie ma wskazań do leczenia supresyjnego) [3].
Rak brodawkowaty tarczycy
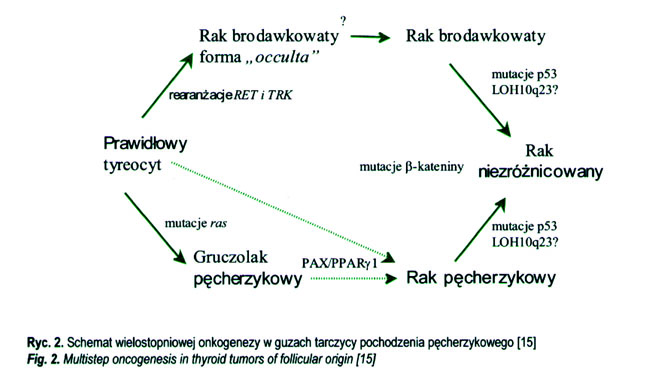
Odmienny jest mechanizm molekularny powstawania nierdzeniastego raka tarczycy wywodzącego się z pojedynczej komórki pęcherzykowej tarczycy (ryc. 2). W literaturze odnotowano występowanie rodzinne raka brodawkowatego tarczycy (PTC), jednak nie udowodniono, aby defekt tylko jednego genu był odpowiedzialny za to zjawisko, tak jak RET onkogen w przypadku MTC. Jednak istnieje kilka genów-kandydatów, które mogą odgrywać rolę w patogenezie rodzinnej postaci PTC, gdzie PTC może być elementem zespołu, w którym występują także raki innych narządów. 5% wszystkich PTC stanowią postaci rodzinne, przy czym uważa się, że typ dziedziczenia jest autosomalny dominujący o niepełnej penetracji [16–17] (tab. II).

Rak pęcherzykowy tarczycy
Rzadko występują formy dziedziczne raka pęcherzykowego tarczycy (FTC), ale właśnie ten typ raka, a nie PTC, występuje znacznie częściej w chorobie Cowdena (PTEN – l Oq23.3).
Rak anaplastyczny tarczycy
Mutacje somatyczne białka p53 są charakterystycznymi zmianami genetycznymi w raku anaplastycznym tarczycy (ATC) [28]. Ciekawy jest fakt, że mutacja genu supresorowego p53 w lini germinatywnej (zespół Le Fraumeni) jest przyczyną powstania mnogich nowotworów (mięsaki, rak piersi przedmenopauzalny, guzy OLTN, białaczki, raki kory nadnerczy), lecz bez obecności ATC [29].
Zespół MEN 1
W skład zespołu wchodzą: (1) nadczynność przytarczyc (85%), (2) guzy wywodzące się z wysp Langerhansa trzustki (75%), (3) guzy przysadki (10-60%) a także, znacznie rzadziej (4) guzy nadnercza, (5) łagodne guzy tarczycy (bardzo rzadko raki) oraz (6) rakowiaki grasicy i oskrzela oraz (7) tłuszczaki. Gen kodujący (MEND ten zespół, zlokalizowany na chromosomie 11, spełnia funkcję genu supresorowego w warunkach prawidłowych. Gen MEN1 koduje białko meninę (z ang. menin), a jego defekt (loss of function) jest odpowiedzialny za wystąpienie cech ww. zespołu [30].
Wrodzone defekty w obrębie komórki pęcherzykowej tarczycy a predyspozycja do nowotworu złośliwego:
a) TPO – 2p24-25 – defekt genu peroksydazy tarczycowej odpowiedzialny za wrodzoną hipotyreozę, wydaje się mieć istotny wpływ na indukcję rozwoju raka pęcherzykowego tarczycy u tych dzieci, gdyż dotyczył wszystkich przypadków dotąd opublikowanych, z wyjątkiem 1 raka brodawkowatego [31–35]. Na podstawie aktualnego stanu wiedzy jest prawdopodobne, że krytyczny region, którego mutacja może zainicjować proces nowotworowy, obejmuje odcinek 2505-2511 w eksonie 14 genu TPO [36–39].
b) PDS – 7g22-31.1 – defekt pendryny (z ang. pendrin) w zespole Pendreda, potwierdzony u pacjenta z inwazyjną formą raka pęcherzykowego z towarzyszącą transformacją anaplastyczną [40].
c) NIS – 19p12 – defekt symportera jodkowo-sodowego, nie został udokumentowany w literaturze odnośnie do predyspozycji do raka tarczycy, ale stwierdzono obniżoną ekspresję tego biała w rakach, szczególnie zaś w raku anaplastycznym (brak ekspresji) [41].
d) Tg – 8g24 – mutacj a genu tyreoglobuliny, odgrywa istotną rolę wśród przyczyn wrodzonej hipotyreozy przebiegającej z wolem, jednakże nie scharakteryzowano dotąd bezpośredniego związku określonej nieprawidłowości genetycznej z rozwojem guza nowotworowego.
e) TSHR – 14g31 – nie udowodniono dotąd, że aktywna mutacja genu dla receptora TSH mogłaby spowodować transformację nowotworową złośliwą w kierunku nierdzeniastego raka tarczycy o podłożu rodzinnym. Ciekawym spostrzeżeniem jest jednak fakt, że w chromosomie 14, w bliskim sąsiedztwie genu TSHR, obecne są inne geny, jak MNG1 (14g32) oraz GD-1 (14g31) [42].
f) PAX-8 – 2g12-q14 – mutacja nawet pojedynczego apelu tego czynnika transkrypcyjnego powoduje wrodzoną hipotyreozę najczęściej z hipoplazją, rzadziej ektopią, co w efekcie nie powoduje wola, a tym samym wola guzkowego [43]. Czynnik ten odpowiedzialny jest za ekspresję genu TPO oraz genu Tg [44].
g) MNG1 (14g31) [45] oraz MNGs (Xp22) [46] wydają się odgrywać znaczącą rolę w powstawaniu rodzinnego wola wieloguzkowego, ale o charakterze nienowotworowym.
Wnioski
Dotychczasowe badania wskazują, że raki wywodzące się z komórek pęcherzykowych maj ą heterogenne podłoże genetyczne w przeciwieństwie do raka rdzeniastego. W grupie raków nierdzeniastych szczególną uwagą należy objąć te rodziny, w których już odnotowano raka tarczycy lub też wystąpiły inne ww. nowotwory, będące elementami zespołu nowotworów mnogich.
Piśmiennictwo
1. Raue F., Kotzerke J., Reinwein D. et al.; Prognostic factors in medullary thyroid carcinoma: evaluation of 741 patients from the German Medullary Thyroid Carcinoma Register; Clin. Invest. 1993:71, 7-12
2. Eng C.; Familial papillary thyroid cancer – urany syndromes, too urany styles; J. Clin. Endocrinol. Metab. 2000:85(5), 1755-1756
3. Gimm 0., Sutter T, Dralle H.; Diagnosis and therapy of sporadic and Familial medullary thyroid carcinoma; J. Cancer Res. Clin. Oncol. 2001:127, 156-165
4. Niedziela M.; Choroba guzkowa tarczycy u dzieci i młodzieży w regionie wielkopolskim – analiza klinicznych i genetycznych czynników występowania nowotworu. Rozprawa habilitacyjna, Akademia Medyczna im. Karola Marcinkowskiego w Poznaniu; Dział Wydawnictw Uczelnianych Akademii Medycznej im. Karola Marcinkowskiego w Poznaniu 2002
5. Gagel R.F.; Polyendocrine disorders: multiple endocrine neoplasia. [w:] Williams Textbook of Endocrinology. Red. Wilson J.D., Foster D.W., Kronenberg H.M., Reed Larsen P. 9th edition; W.B. Saunders Company Philadelphia 1998, 1627-1649
6. Holmes J.M., Engel J.M., Ticho B.H et al.:; Syndromes with ophthalmic manifestations. [w:] Pediatric ophthalmology and strabismus. Red. Wright K.W.; Mosby 1995, 714-716
7. Mulligan L.M., Marsh D.J., Robinson B.G. et al.; Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the Intemational RET Mutation Consortium; J. Intem. Med. 1995:238, 343-346
8. Eng C., Clayton D., Scuffenecker I. et al.; The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis; J. Am. Med. Assoc. 1996:276, 1575-1579
9. Donis-Keller H., Dou S., Chi D. et al.; Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC; Hum. Mol. Genet. 1993:2, 851-856
10. Mulligan L.M., Kwok J.B., Healey C.S. et al.; Germ-line mutations of the RET proto-oncogene in multiple endocrine type 2A; Nature 1993:363, 458-460
11. Gordon C.M., Majzoub J.A., Marsh D.J. et al.; Four cases of mucosal neuroma syndromes multiple endocrine neoplasm 2B or not 2B; J. Clin. Endocrinol. Metab. 1998:83(1),17-20
12. Offit K., Biesecker B.B., Burt R.W. et al.; Statement of the American Socjety of Clinical Oncology – genetic testing for cancer susceptibility; J. Clin. Oncol. 1996:14,1730-1736
13. Wohlik N., Cote G.J., Evans D.B. et al.; Application of genetic screening information to the management of medullary thyroid carcinoma and multiple endocrine neoplasia type 2; Endocrinol. Metab. Clin. North Am. 1996:25, 1-25
14. Wiench M., Wygoda Z., Gubala E. et al.; Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients; J. Clin. Oncol. 2001:19(5), 1374-1380
15. Gimm O.; Thyroid cancer; Cancer letters 2001:163, 143-156
16. Malchoff C.D., Malchoff D.M.; The genetics of hereditary nonmedullary thyroid carcinoma; J. Clin. Endocrinol. Metab. 2002:87(6), 2455-2459
17. Fagin J.A.; Familial nonmedullary thyroid carcinoma – the case for genetic susceptibility; J. Clin. Endocrinol. Metab. 1997:82(2), 342-344
18. Malchoff C.D., Sarfarazi M.S., Tendler B. et al.; Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome; J. Clin. Endocrinol. Metab. 2000:85, 1758-1764
19. Canzian F., Amati P., Harach R. et al.; A gene predisposing to familial thyroid tumors with celt oxyphilia maps to chromosome 19p13.2; Am. J. Hum. Genet. 1998:63, 1743-1748
20. Bevan S., Pal T., Greenberg C.R. et al.; A comprehensive analysis of MNG1, TC01, fPTC, PTEN, TSHR and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1; J. Clin. Endocrinol. Metab. 2001:86(8), 3701-3704
21. Cohen A.J., Li F.P., Berg S. et al.; Hereditary renal cell carcinoma asociated with chromosomel translocation; N. Engl. J. Med. 1997:301, 592-595
22. Plail R.O., Bussey H.J.R., Glazer G., Thomson J.P.S.; Adenomatous polyposis: an association with carcinoma of the thyroid; Br. J. Surg. 1987:74, 377-380
23. Bell B., Mazzaferri E.L.; Familial adenomatous polyposis (Gardner’s syndrome) and thyroid carcinoma. A case report and review of the literature; Dig. Dis. Sci. 1993:38, 185-190
24. Kinzler K.W., Nilbert M.C., Su N.K.L.; Identification of FAP locus genes from chromosome 5g21; Science 1991:253, 661-664
25. Mallory S.B.; Cowden syndrome (multiple hamartoma syndrome); Dermatol. Clin. 1995:13, 27-31
26. Liaw D., Marsh D.J., Li J. et al.; Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome; Nat. Genet. 1997:16, 64-67
27. McKay J.D., Lesueur F., Jonard L. et al.; Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2g21; Am. J. Hum. Genet. 2001:69, 440-446
28. Fagin J.A., Matsuo K., Karmarkar A. et al.; High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas; J. Clin. Invest. 1992:91, 179-184
29. Levine A.; p53, the cellular gatekeeper for growth and division; Cell 1997:88, 323-331
30. Larsson C., Skogseid B., Oberg K., Nakamura Y.; Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma; Nature 1988:332, 85-87
31. Morris H.P., Dalton A.J., Green G.D.; Malignant thyroid tumors occurring in the mouse after prolonged hormonal unbalance during the ingestion of thiouracil; J. Clin. Endocrinol. Metab. 1951:11, 1281-1285
32. Medeiros-Neto G., Stanbury J.B.; Inherited disorders of the thyroid system; CRC Press, Boca Raton 1994, 207-218
33. Yashiro T., Ito K., Akiba M. et al.; Papillary carcinoma of the thyroid arising from dyshormonogenetic goiter; Endocrinol. Jpn. 1987:34, 955-964
34. Cooper D.S., Axelrod L., De Groot L.J. et al.; Congenital goiter and the development of metastatic follicular carcinoma with evidence for a leak of nonhormonal iodide: clinical, pathological, kinetic, and biochemical studies and a review of the literature; J. Clin. Endocrinol. Metab. 1981:52, 294-306
35. Crooks J., Greig W.R., Branwood A.W.; Dyshormonogenesis and carcinoma of the thyroid gland; Scot. Med. J. 1963:8, 303
36. Medeiros-Neto G., Gil-da-Costa M.J., Santos C.L.S. et al.; Metastatic thyroid carcinoma arising from congenital goiter due to mutation in the thyroperoxidse gene; J. Clin. Endocrinol. Metab. 1998:83, 4162-4166
37. Kotani T., Umeki K., Yamamoto I. et al.; A novel mutation in the human thyroid peroxidase gene resulting in a total iodide organification defect; J. Endocrinol. 1999:160, 267-273
38. Kotani T., Umeki K., Yamamoto I. et al.; lodide organification defects resulting from cosegregation of mutated and null thyroid peroxidase alleles; Mol. Cell. Endocrinol. 2001:182(1), 61-68
39. Niedziela M., Ambrugger P., Krude H. et al.; TPO gene as a candidate gene in the pathogenesis of thyroid neoplasia of patients with congenital hypothyroidism; J. Endocrinol. Invest. 2001:24, suppl. 6, 431.2
40. Camargo R., Limbert E., Gillam M. et al.; Aggressive metastatic follicular thyroid carcinoma with anaplastic transformation arising from a long-standing goiter in a patient with Pendred’s syndrome; Thyroid 2001:11(10), 981-988
41. Czamocka B., Pastuszka D., Lyczkowska A. et al.; lodide symporter and thyroid peroxidase (TPO) expression in thyroid epithelial cancer; J. Endocrinol. Invest. 2001:24, suppl. 6, 48
42. Tomer Y, Barbesino G., Greenberg D.A. et al.; Linkage analysis of candidate genes in autoimmune thyroid disease. III. Detailed analysis of chromosome 14 localizes Graves’ Disease-1 (GD-1) close to multinodular goiter-1 (MNG-1); J. Clin. Endocrinol. Metab. 1998:83(12), 4321-4327
43. Macchia P.E., Lapi P., Krude K. et al.; PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis; Nat. Genet. 1998:19, 83-86
44. Zannini M., Francis-Lang H., Plachov D., Di Lauro R.; Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homedomain and activates transcription from two thyroid-specific promoters; EMBO J. 1992:12, 4230-4241
45. Bignell G.R.: Canzian F., Shayeghi M. et al.; Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer; Am. J. Hum. Genet. 1997:61, 1123-1130
46. Capon F., Tacconelli A, Giardina E.; Mapping a dominant form of multinodular goiter to chromosome Xp22; Am. J. Hum. Genet. 2000:67,1004-1007








